Дарбэстим — инструкция по применению
Синонимы, аналоги
Статьи
Внимательно прочтите эту инструкцию перед тем, как начать прием/использование этого лекарственного средства.
Сохраните инструкцию, она может потребоваться вновь.
Если у Вас возникли вопросы, обратитесь к врачу.
Это лекарственное средство назначено лично Вам, и его не следует передавать другим лицам, поскольку оно может причинить им вред даже при наличии тех же симптомов, что и у Вас.
Регистрационный номер:
ЛП-005411
Торговое наименование:
Дарбэстим®
Международное непатентованное наименование (МНН):
Дарбэпоэтин альфа (darbepoetin alfa)
Лекарственная форма:
раствор для инъекций.
Состав:
Действующее вещество в одном предварительно заполненном шприце Дарбэпоэтин альфа (рекомбинантный): 10 мкг (25 мкг/мл), 15 мкг (40 мкг/мл), 20 мкг (40 мкг/мл), 30 мкг (100 мкг/мл), 40 мкг (100 мкг/мл), 50 мкг (100 мкг/мл), 60 мкг (200 мкг/мл), 80 мкг (200 мкг/мл), 100 мкг (200 мкг/мл), 150 мкг (500 мкг/мл), 300 мкг (500 мкг/мл), 500 мкг (500 мкг/мл).
Вспомогательные вещества в 1 мл раствора: натрия дигидрофосфата моногидрат – 2,118 мг, натрия гидрофосфат – 0,661 мг, натрия хлорид 8,182 мг, полисорбат 80 – 0,05 мг, вода для инъекций – до 1,0 мл.
Описание:
Прозрачная бесцветная жидкость.
Фармакотерапевтическая группа:
Гемопоэза стимулятор (антианемическое средство).
ATX:
В03ХА02
Фармакологические свойства
Дарбэпоэтин альфа производится с использованием генной технологии в клетках яичников китайского хомяка (СНО-К1).
Фармакодинамика
Дарбэпоэтин альфа стимулирует эритропоэз по тому же механизму, что и эндогенный эритропоэтин. Дарбэпоэтин альфа содержит пять N-связанных углеводных цепей, в то время как эндогенный гормон и рекомбинантные человеческие эритропоэтины (рчЭпо) имеют всего три цепи. Дополнительные остатки сахаров, с молекулярной точки зрения, не отличаются от таковых, представленных в эндогенном гормоне. Вследствие повышенного содержания углеводов дарбэпоэтин альфа обладает более длительным периодом полувыведения в сравнении с рчЭпо, а, следовательно, и большей активностью in vivo. Несмотря на указанные изменения молекулярной структуры дарбэпоэтин альфа сохраняет очень узкую специфичность к эритропоэтиновому рецептору.
Эритропоэтин – фактор роста, который в основном стимулирует образование эритроцитов. Рецепторы к эритропоэтину могут экспрессироваться на поверхности различных опухолевых клеток.
Больные с хронической почечной недостаточностью
В 2 клинических исследованиях было выявлено, что у пациентов с ХПН риск летального исхода и серьезных сердечно-сосудистых нежелательных явлений выше при применении стимуляторов эритропоэза до более высоких целевых уровней гемоглобина при сравнении с более низкими (135 г/л (8,4 ммоль/л) против 113 г/л (7,1 ммоль/л); 140 г/л (8,7 ммоль/л) против 100 г/л (6,2 ммоль/л).
В рандомизированном, двойном слепом коррекционном исследовании (n = 358) сравнения режимов дозирования один раз каждые две недели и один раз в месяц у пациентов с хронической почечной недостаточностью, не находящихся на диализе, применение дарбэпоэтина альфа для коррекции анемии один раз в месяц было не хуже, чем один раз каждые две недели. Среднее время (1-й Квартиль, 3-й Квартиль) достижения коррекции уровня гемоглобина (≥100 г/л и ≥10 г/л по сравнению с исходным уровнем) составило 5 недель для обоих режимов дозирования (3,7 недель для режима дозирования один раз каждые две недели и 3,9 недель для режима дозирования один раз в месяц). В период оценки (недели 29-33), среднее значение (95% ДИ) эквивалентной дозы составило 0,20 (0,17; 0,24) мкг/кг при режиме дозирования один раз каждые две недели и 0,27 (0,23; 0,32) мкг/кг при режиме дозирования один раз в месяц.
В рандомизированном, двойном слепом, плацебо-контролируемом исследовании (TREAT) 4038 пациентов с хронической почечной недостаточностью, с диабетом 2 типа и уровнем гемоглобина ≤110 г/л, не находящихся на диализе, получали дарбэпоэтин альфа с целью достижения уровня гемоглобина 130 г/л или плацебо (с назначением дарбэпоэтина альфа при снижении уровня гемоглобина ниже 90 г/л). Исследование не достигло основной цели, заключающейся в снижении риска смертности по любым причинам или по сердечно-сосудистой заболеваемости (дарбэпоэтин альфа vs плацебо; соотношение рисков 1,05; 95% ДИ (0,94; 1.17), равно как и цели заключающейся в снижении смертности по любым причинам и прогрессирования до терминальной стадии почечной недостаточности (ТСПН) (дарбэпоэтин альфа vs плацебо; соотношение рисков 1,06; 95% ДИ (0,95; 1,19). Анализ индивидуальных компонентов композитных конечных точек показал следующее соотношение рисков (95% ДИ): летальный исход 1,05 (0,92; 1,21), хроническая сердечная недостаточность (ХСН) 0,89 (0,74; 1,08), инфаркт миокарда (ИМ) 0,96 (0,75; 1,23), инсульт 1,92 (1,38; 2,68), госпитализация в связи с ишемией миокарда 0,84 (0,55; 1,27), ТСПН 1,02 (0,87; 1.18).
Онкологические больные, получающие химиотерапию
Выживаемость и прогрессирование опухоли были изучены в общей сложности у 2833 пациентов в рамках пяти крупных контролируемых исследований. Из них четыре были двойными слепыми и плацебо-контролируемыми, а одно – открытым. В двух исследованиях включались больные, которым уже было проведено химиотерапевтическое лечение. В двух исследованиях целевой уровень гемоглобина устанавливался равным и выше 130 г/л, а в трех других – в интервале от 120 до 140 г/л. В открытом исследовании не получено различий в показателях общей выживаемости между группой, получавшей лечение рчЭПО, и контрольной. В четырех плацебо-контролируемых исследованиях показатели риска были в пользу контроля и находились в пределах от 1,25 до 2,47. В этих четырех исследованиях был выявлен необъяснимый статистически достоверный прирост смертности по сравнению с контролем у больных с типичными видами рака и анемией, лечение которой проводилось рчЭПО. Сравнение частоты тромбозов и других осложнений в группах, получавших лечение рчЭПО, и контрольной, не дает удовлетворительного объяснения причин этого прироста.
Также был проведен систематический анализ 57 исследований, включавших суммарно более 9000 онкологических пациентов. При метаанализе общей выживаемости показатель риска равнялся 1,08 в пользу контроля (ДИ 95%: 0,99-1,18; 8167 пациентов в 42 исследованиях).
У пациентов, получавших лечение рчЭПО, отмечалось повышение относительного риска развития тромбоэмболических событий (ОР=1,67; ДИ 95%: 1,35-2,06; 6769 пациентов в 35 исследованиях). Таким образом, существует достаточный объем данных, свидетельствующих о возможности возникновения значительного вреда при лечении онкологических больных рчЭПО. Неясно, до какой степени это применимо к случаям назначения рекомбинантных человеческих эритропоэтинов для достижения целевого уровня гемоглобина менее 130 г/л у пациентов с онкологическими заболеваниями, которые получают химиотерапию, поскольку в проанализированных данных имелось незначительное число пациентов с такими характеристиками.
Также был проведен анализ данных более чем у 13900 пациентов со злокачественными заболеваниями (химиотерапия, лучевая терапия, химиотерапия и лучевая терапия или отсутствие терапии), включенных в 53 контролируемых клинических исследования нескольких эпоэтинов. Метаанализ данных по общей выживаемости выявил соотношение рисков 1,06 в пользу контрольной группы (95% ДИ: 1,00; 1,12; 53 исследования и 13933 пациента), и для пациентов со злокачественными заболеваниями, получающими химиотерапию, соотношение рисков общей выживаемости составило 1,04 (95% ДИ: 0,97; 1,11; 38 исследований и 10441 пациента). Метаанализ также указывает на значительное повышение относительного риска тромбоэмболических событий у пациентов со злокачественными образованиями, получающих рекомбинантный человеческий эритропоэтин (см. раздел «Особые указания»).
Доклинические данные по безопасности
Во всех исследованиях на крысах и собаках при применении дарбэпоэтина альфа значимо возрастала концентрация гемоглобина, гематокрита, эритроцитов и ретикулоцитов, что соответствует ожидаемому фармакологическому эффекту. Нежелательные явления при введении очень высоких доз препарата рассматривались как следствие усиленного фармакологического действия (снижения тканевого кровотока вследствие увеличения вязкости крови). Сюда же были отнесены миелофиброзы и гипертрофия селезенки, а также расширение комплекса QRS на ЭКГ у собак, без нарушения сердечного ритма и влияния на интервал QT.
Дарбэпоэтин альфа не обладал каким-либо генотоксическим потенциалом и не оказывал влияния на пролиферацию клеток негематологического ряда ни in vitro, ни in vivo. В исследованиях по хронической токсичности не наблюдалось туморогенного или неожиданного митогенного ответа ни в одном изученном типе тканей. В продолжительных исследованиях на животных оценка канцерогенного потенциала дарбэпоэтина альфа не выполнялась.
В испытаниях, проводившихся на крысах и кроликах, не наблюдалось клинически значимого влияния на беременность, эмбриональное/фетальное развитие, роды или постнатальное развитие. Уровень проникновения препарата через плаценту был минимальным. Изменений фертильности не отмечалось.
Фармакокинетика
В связи с повышенным содержанием углеводов концентрация циркулирующего в крови дарбэпоэтина альфа превышает минимальную концентрацию необходимую для стимуляции эритропоэза в течение более продолжительного времени в сравнении с эквивалентными дозами рчЭпо, что позволяет снизить частоту введения дарбэпоэтина альфа с сохранением эквивалентного уровня биологического ответа.
Больные с хронической почечной недостаточностью
Фармакокинетика дарбэпоэтина альфа была изучена у больных с хронической почечной недостаточностью при внутривенном и подкожном введении препарата. Его период полувыведения составлял 21 час (стандартное отклонение (СО) 7,5) при внутривенном введении. Клиренс дарбэпоэтина альфа составил 1,9 мл/час/кг (СО 0,56), а объем распределения (Орс) был приблизительно эквивалентен объему плазмы (50 мл/кг). При подкожном введении препарата его биодоступность соответствовала 37%. При ежемесячном подкожном введении дарбэпоэтина альфа в дозе от 0,6 до 2,1 мкг/кг его период полувыведения составлял 73 часа (СО 24). Более продолжительный период полувыведения дарбэпоэтина альфа при подкожном введении, по сравнению с внутривенным, обусловлен кинетикой абсорбции. В ходе клинических исследований минимальное накопление препарата наблюдалось при любом способе введения. В доклинических исследованиях было продемонстрировано, что почечный клиренс дарбэпоэтина минимален (до 2% общего клиренса) и не оказывает влияния на период полувыведения препарата из сыворотки.
Фармакокинетика дарбэпоэтина альфа изучалась у детей (3-16 лет) с ХПН, находящихся или не находящихся на диализе, при этом забор образцов проводился от момента однократного подкожного или внутривенного введения препарата до одной недели (168 часов) после введения. Периоды забора образцов были такой же продолжительности, как и у взрослых с ХПН, и сравнение показало, что фармакокинетика дарбэпоэтина альфа у взрослых и детей с ХПН похожа. После внутривенного введения препарата отмечалось приблизительно 25% различие между взрослыми и детьми в отношении площади под фармакокинетической кривой «концентрация-время» от нулевой отметки времени до бесконечности (AUC[0-∞]); тем не менее, указанное различие для детей составило менее двухкратного диапазона AUC[0-∞]. После подкожного введения препарата величина AUC[0-∞] у взрослых и детей была аналогичной. Как после внутривенного, так и после подкожного введения препарата, период полувыведения препарата у детей и взрослых с ХПН был сходен.
Онкологические больные, получающие химиотерапию
После подкожного введения препарата в дозе 2,25 мкг/кг взрослым онкологическим больным средние максимальные концентрации дарбэпоэтина альфа, составляющие 10,6 нг/мл (СО 5,9), устанавливались в среднем в течение 91 часа (СО 19,7). Эти параметры соответствовали линейной фармакокинетике дозы в широком диапазоне значений (от 0,5 до 8 мкг/кг при еженедельном введении от 3 до 9 мкг/кг при введении раз в две недели).
Фармакокинетические параметры не изменялись при многократном дозировании в течение 12 недель (еженедельное введение или введение раз в две недели). Отмечалось ожидаемое умеренное повышение (<2-кратное) сывороточной концентрации препарата при достижении равновесного состояния, но не было выявлено признаков его накопления при повторном назначении. Исследования фармакокинетики были выполнены с привлечением пациентов с индуцированной во время химиотерапии анемией, которые в комбинации с химиотерапией подкожно получали инъекции дарбэпоэтина альфа в дозе 6,75 мкг/кг раз в три недели. В данном исследовании среднее значение (СО) периода полувыведения составляло 74 (СО 27) часа.
Показания к применению
- Лечение симптоматической анемии у взрослых и детей, страдающих хронической почечной недостаточностью (ХПН).
- Терапия симптоматической анемии у взрослых онкологических больных с немиелоидными злокачественными новообразованиями, получающих химиотерапию.
Противопоказания
- Повышенная чувствительность к дарбэпоэтину альфа, рчЭпо или к любому компоненту препарата.
- Неконтролируемая артериальная гипертензия.
С осторожностью
- Заболевания печени; серповидно-клеточная анемия; эпилепсия.
Применение в период беременности и грудного вскармливания
Полноценные, контролируемые исследования дарбэпоэтина альфа у беременных женщин не проводились.
В экспериментах на животных не было продемонстрировано прямого повреждающего действия препарата на течение беременности, на эмбриональное/фетальное развитие, на роды или постнатальное развитие. Влияния на фертильность выявлено не было. При назначении препарата беременным женщинам следует соблюдать осторожность.
Неизвестно, экскретируется ли дарбэпоэтин альфа в грудное молоко. Поэтому не исключен риск для ребенка при грудном вскармливании. Принятие решения о прекращении грудного вскармливания или прекращении/отмене лечения препаратом Дарбэстим® следует принимать, учитывая пользу грудного вскармливания для ребенка и пользу лечения для матери.
Способ применения и дозы
Лечение дарбэпоэтином альфа должно проводиться врачами, имеющими опыт назначения по вышеупомянутым показаниям.
Дарбэстим® поставляется готовым для применения в предварительно заполненных шприцах (ПЗШ). Инструкции по применению препарата, обращению с ним и порядку его уничтожения приводятся в разделе «Особые указания по использованию».
Терапия симптоматической анемии в сочетании с хронической почечной недостаточностью (ХПН) у взрослых и детей
Симптомы анемии и последствия могут варьировать в зависимости от возраста пациентов, их пола и тяжести заболевания; в каждом случае необходим анализ индивидуальных клинических данных пациента лечащим врачом.
Дарбэпоэтин альфа может применяться подкожно или внутривенно для повышения уровня гемоглобина, но не выше 120 г/л. У больных, не находящихся на гемодиализе, подкожный способ введения является предпочтительным, так как позволяет избежать пункций периферических вен.
Уровень гемоглобина у пациентов подвержен индивидуальным колебаниям, в том числе иногда выше или ниже желаемых целевых значений. При отклонении уровня гемоглобина за пределы целевых значений проводят модификацию дозы, при этом под целевым значением следует рассматривать интервал от 100 до 120 г/л. Следует избегать стойкого повышения уровня гемоглобина выше 120 г/л, указания по коррекции дозы при повышении уровня гемоглобина выше 120 г/л представлены ниже. Также следует избегать повышения уровня гемоглобина более чем на 20 г/л за 4-недельный период. В этом случае также необходима коррекция дозы.
Лечение дарбэпоэтином альфа включает две стадии – фаза коррекции и поддерживающая фаза. Рекомендации по применению и дозированию у взрослых и детей в инструкции приводятся отдельно. Применение у детей в возрасте меньше 1 года не изучалось.
Взрослые пациенты с хронической почечной недостаточностью
Фаза коррекции
Начальная доза при подкожном или внутривенном введении должна составлять 0,45 мкг/кг массы тела при однократном еженедельном введении. Альтернативно, для больных, не получающих диализ, следующие начальные дозы препарата также могут назначаться подкожно: 0,75 мкг/кг массы тела каждые две недели или 1,5 мкг/кг массы тела один раз в месяц. Если повышение концентрации гемоглобина оказывается недостаточным (менее 10 г/л за 4 недели), доза препарата увеличивается приблизительно на 25%. Повышение дозы препарата не должно осуществляться чаще, чем один раз в четыре недели.
Если увеличение содержания гемоглобина превышает 20 г/л за 4 недели, дозу препарата следует уменьшить примерно на 25%. В случае, когда уровень гемоглобина превышает 120 г/л, следует рассмотреть возможность уменьшения дозы препарата. Если содержание гемоглобина продолжает увеличиваться, дозу следует снизить примерно на 25%. Если после снижения дозы, гемоглобин продолжает повышаться, необходимо временно прекратить применение препарата до начала снижения уровня гемоглобина, после чего можно возобновить терапию, причем дозу препарата следует уменьшить примерно на 25% от предыдущей дозы.
Гемоглобин следует измерять еженедельно или раз в две недели до его стабилизации. В последующем промежутки между измерениями гемоглобина можно увеличить.
Поддерживающая фаза
У пациентов, находящихся на диализе, можно продолжить вводить препарат Дарбэстим® один раз в неделю или перейти на введение один раз каждые две недели. При переводе пациентов, находящихся на диализе, с еженедельных инъекций на режим введения однократно раз в две недели, исходная доза должна вдвое превышать дозу, вводившуюся один раз в неделю. Для пациентов, не получающих диализа, можно продолжить вводить препарат Дарбэстим® один раз в неделю или один раз каждые две недели или один раз в месяц. Для пациентов, получающих препарат Дарбэстим® один раз каждые две недели, после достижения требуемой концентрации гемоглобина, его подкожное введение может производиться один раз в месяц с использованием исходной дозы вдвое превышающей предыдущую дозу, вводившуюся раз в две недели.
Титрование дозы с целью поддержания требуемой концентрации гемоглобина следует производить так часто, как это требуется.
Если для поддержания требуемого гемоглобина необходима оптимизация дозы препарата Дарбэстим®, ее рекомендуется увеличивать приблизительно на 25%. В случае, если наблюдается повышение гемоглобина более, чем 20 г/л за 4 недели, дозу препарата следует уменьшить приблизительно на 25%, в зависимости от скорости повышения. Если содержание гемоглобина превышает 120 г/л, следует рассмотреть возможность уменьшения дозировки препарата. Если содержание гемоглобина продолжает увеличиваться, дозу следует снизить примерно на 25%. Если после снижения дозы, гемоглобин продолжает повышаться, необходимо временно прекратить применение препарата до начала снижения уровня гемоглобина, после чего можно возобновить терапию, причем дозировку препарата следует уменьшить примерно на 25% от предыдущей дозы.
Следует проводить тщательное наблюдение за пациентами для обеспечения адекватной коррекции анемии с применением минимальных одобренных доз препарата Дарбэстим®.
После любого изменения дозы или режима введения, содержание гемоглобина следует контролировать каждую 1 или 2 недели. Изменение дозы во время поддерживающей фазы должно выполняться не чаще одного раза в две недели.
При изменении пути введения препарата следует использовать те же дозы препарата и осуществлять мониторинг концентрации гемоглобина раз в 1-2 недели с целью поддержания требуемого уровня гемоглобина.
Взрослых пациентов, получающих еженедельно по одной, две или три инъекции рчЭпо, можно перевести на режим однократного еженедельного введения препарата Дарбэстим® или его введение один раз в две недели. Исходную еженедельную дозу препарата Дарбэстим® (мкг/неделю) определяют, разделив общую еженедельную дозу рчЭпо (МЕ/неделю) на 200. Исходную дозу препарата Дарбэстим® (мкг/ в две недели) при режиме введения один раз в две недели определяют путем деления суммарной кумулятивной дозы рчЭпо, введенного за двухнедельный период, на 200. Ввиду известной индивидуальной вариабельности, для отдельных больных может потребоваться титрование доз до получения оптимального терапевтического эффекта. При замещении рчЭпо на препарат Дарбэстим® измерение уровня гемоглобина следует выполнять не реже одного раза в неделю или в две недели, а способ введения препарата должен оставаться неизменным.
Дети с хронической почечной недостаточностью
Фаза коррекции
Для детей в возрасте 11 лет и старше начальная доза при подкожном или внутривенном введении препарата составляет 0,45 мкг/кг веса тела в виде однократной инъекции один раз в неделю. У пациентов, не получающих диализ, может применяться начальная доза, равная 0,75 мкг/кг, подкожно один раз в две недели. Если увеличение уровня гемоглобина недостаточно (менее 10 г/л за 4-недельный период), необходимо увеличить дозу препарата примерно на 25%. Увеличение дозы должно проводиться не чаще одного раза в четыре недели.
Если увеличение содержания гемоглобина превышает 20 г/л за 4 недели, дозу препарата следует уменьшить примерно на 25% в зависимости от степени увеличения уровня гемоглобина. В случае, когда уровень гемоглобина превышает 120 г/л, следует рассмотреть возможность уменьшения дозы препарата. Если содержание гемоглобина продолжает увеличиваться, дозу следует снизить примерно на 25%. Если после снижения дозы, гемоглобин продолжает повышаться, необходимо временно прекратить применение препарата до начала снижения уровня гемоглобина, после чего можно возобновить терапию, причем дозу препарата следует уменьшить примерно на 25% от предыдущей дозы. Гемоглобин следует измерять еженедельно или раз в две недели до его стабилизации. В последующем промежутки между измерениями гемоглобина можно увеличить.
Лечение анемии препаратом Дарбэстим® в фазе коррекции у детей в режиме дозирования один раз в месяц не изучено.
Рекомендаций касательно коррекции уровня гемоглобина у детей в возрасте от 1 года до 10 лет нет.
Поддерживающая фаза
У детей 11 лет и старше в поддерживающую фазу терапии введение препарата Дарбэстим® можно продолжать в режиме один раз в неделю или один раз в две недели. Пациенты, находящиеся на диализе, при переводе их с режима дозирования препарата Дарбэстим® один раз в неделю в режим один раз в две недели первоначально должны получать дозу, эквивалентную удвоенной при однократном в неделю режиме введения. Если пациент не находится на диализе, после того, как достигнут целевой уровень гемоглобина в режиме дозирования препарата 1 раз в две недели, препарат Дарбэстим® может назначаться подкожно 1 раз в месяц, при этом начальная дозировка должна составлять удвоенную дозу от той, что использовалась в режиме 1 раз в две недели.
Для детей в возрасте от 1 года до 18 лет клинические данные показали, что пациенты, получающие рчЭпо два или три раза в неделю, могут быть переведены на препарат Дарбэстим®, вводимый 1 раз в неделю, и пациенты, получающие рчЭпо один раз в неделю, могут быть переведены на режим введения один раз в две недели. Начальная дозировка препарата Дарбэстим® для детей (мкг/нед), вводимого еженедельно может быть определена путем деления суммарной недельной дозы рчЭпо (МЕ/нед) на 240. Начальная дозировка препарата Дарбэстим® при введении каждые 2 недели (мкг/каждые 2 недели) может быть определена путем деления суммарной дозы рчЭпо за двухнедельный период на 240. По причине индивидуальных различий для отдельных пациентов требуется подбор оптимальной терапевтической дозы. При замене рчЭпо на препарат Дарбэстим®, уровень гемоглобина должен контролироваться каждые 1-2 недели, и при этом должен использоваться один и тот же способ введения препарата.
Титрование дозы с целью поддержания требуемой концентрации гемоглобина следует производить так часто, как это требуется.
Если для поддержания требуемого гемоглобина необходима оптимизация дозы препарата Дарбэстим®, ее рекомендуется увеличивать приблизительно на 25%. Если увеличение содержания гемоглобина превышает 20 г/л за 4 недели, дозу препарата следует уменьшить примерно на 25% в зависимости от степени увеличения уровня гемоглобина. В случае, когда уровень гемоглобина превышает 120 г/л, следует рассмотреть возможность уменьшения дозы препарата. Если содержание гемоглобина продолжает увеличиваться, дозу следует снизить примерно на 25%. Если после снижения дозы, гемоглобин продолжает повышаться, необходимо временно прекратить применение препарата до начала снижения уровня гемоглобина, после чего можно возобновить терапию, причем дозу препарата следует уменьшить примерно на 25% от предыдущей дозы.
Состояние пациентов следует тщательно мониторировать, для уверенности, что используемые минимальные одобренные дозы препарата Дарбэстим® обеспечивают адекватный контроль симптомов анемии.
После любого изменения дозы или режима введения, содержание гемоглобина следует контролировать каждую 1 или 2 недели. Изменение дозы во время поддерживающей фазы должно выполняться не чаще одного раза в две недели.
При изменении пути введения препарата следует использовать те же дозы препарата и осуществлять мониторинг концентрации гемоглобина раз в 1-2 недели с целью поддержания требуемого уровня гемоглобина.
Лечение симптоматической анемии, индуцированной химиотерапией, у пациентов с онкологическими заболеваниями
У пациентов с анемией (например, при концентрации гемоглобина равной или ниже 100 г/л) препарат Дарбэстим® может применяться подкожно для повышения уровня гемоглобина, но не выше 120 г/л. Симптоматика и последствия анемии зависят от возраста пациентов, их пола и тяжести заболевания. В каждом случае необходим анализ индивидуальных клинических данных пациента лечащим врачом.
Поскольку содержание гемоглобина в крови – индивидуальный показатель, для которого характерно выраженное разнообразие, у некоторых пациентов его содержание может как превышать целевой уровень, так и быть меньше его. В этом случае помогает коррекция дозировки препарата, с учетом того, что целевой уровень гемоглобина составляет от 100 г/л до 120 г/л. Следует избегать повышения концентрации гемоглобина более 120 г/л; ниже представлено руководство по коррекции дозы в случае, если содержание гемоглобина превышает 120 г/л.
Рекомендованная начальная доза препарата – 500 мкг (6,75 мкг/кг) 1 раз в 3 недели либо по 2,25 мкг/кг 1 раз в неделю. Если клинический ответ (утомляемость, содержание гемоглобина) через девять недель неадекватен, дальнейшая терапия может оказаться неэффективной. Применение препарата Дарбэстим® прекращают примерно через четыре недели после завершения химиотерапии.
После достижения целевого уровня гемоглобина дозировку препарата следует уменьшить на 25-50%, для адекватного контроля симптоматики анемии с использованием минимальных одобренных доз препарата Дарбэстим®. Возможно титрование дозы между 500 мкг, 300 мкг и 150 мкг.
Следует производить тщательный мониторинг состояния пациентов. Если уровень гемоглобина у пациента превышает 120 г/л, дозу препарата следует уменьшить на 25-50%. Если содержание гемоглобина превышает 130 г/л, следует временно прекратить применение препарата Дарбэстим®. После снижения уровня гемоглобина до 120 г/л или ниже, терапию можно возобновить, дозировка препарата при этом должна быть примерно на 25% меньше предыдущей.
Если увеличение уровня гемоглобина превышает 20 г/л за 4 недели, следует уменьшить дозировку препарата на 25-50%.
Техника процедуры инъекции препарата Дарбэстим® в предварительно заполненных шприцах или в предварительно заполненных шприцах с защитным устройством для иглы
Этот раздел описывает процедуру инъекции препарата Дарбэстим®, которую вы можете выполнить самостоятельно. Перед началом применения препарата в предварительно заполненных шприцах и предварительно заполненных шприцах с защитным устройством для иглы, пожалуйста, сначала ознакомьтесь с «Общими рекомендациями», приведенными ниже (раздел 1), а затем, с рекомендациями, приведенными в разделе 2.
Раздел 1. Общие рекомендации
Очень важно, чтобы Вы не делали инъекцию сами, пока ваш лечащий врач, медицинская сестра или провизор не научит Вас. Если у Вас будут вопросы, то проконсультируйтесь с Вашим врачом, медицинской сестрой.
Перед началом инъекции
Внимательно прочитайте все рекомендации перед введением препарата.
Как Вам или тому, кто делает вам эту инъекцию, использовать ПЗШ?
Ваш врач назначил Вам препарат Дарбэстим® для подкожных инъекций. Ваш лечащий врач, медицинская сестра или провизор расскажет Вам какое количество препарата и как часто необходимо вводить.
Оборудование
Для самостоятельных инъекций вам потребуется:
- Один шприц (ПЗШ)/ ПЗШ с защитным устройством для иглы с препаратом Дарбэстим®;
- Спиртовая салфетка.
Перед проведением инъекции
- Достать шприц из холодильника. Для извлечения предварительно заполненного шприца из пачки, возьмитесь за центр прозрачного защитного устройства для иглы.
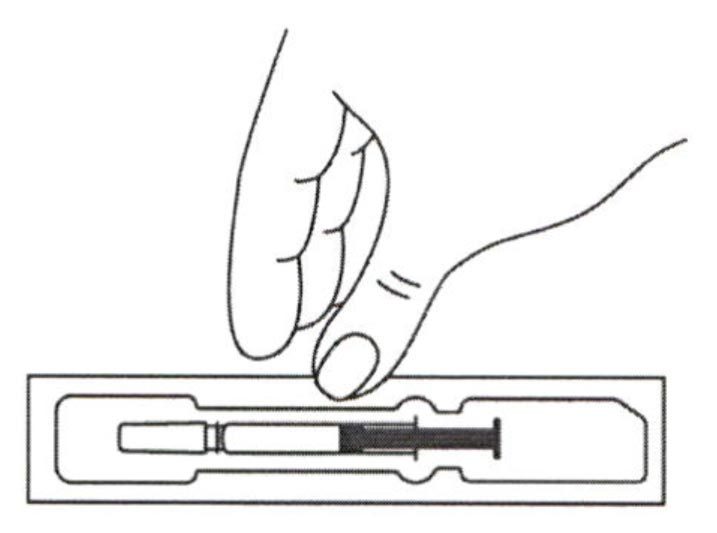
- Оставить шприц при комнатной температуре примерно на 30 минут. Это позволит сделать инъекцию более комфортной.
- Пожалуйста, обратите внимание на следующие инструкции:
- Не подогревать предварительно заполненный шприц в горячей воде или в микроволновой печи.
- Не оставлять заполненный шприц под солнечными лучами.
- Не брать заполненный шприц за шток поршня, серый колпачок иглы или усики защитного устройства для иглы.
- Не встряхивать шприц.
- Не удалять колпачок предварительно заполненного шприца до тех пор, пока Вы не готовы к инъекции
- Не пытайтесь активировать предварительно заполненный шприц перед инъекцией.
- He пытайтесь снять прозрачное защитное устройство для иглы с предварительно заполненного шприца.
Хранить предварительно заполненные шприцы в недоступном для детей месте.
- Перед использованием необходимо проверить следующее:
- Вы используете тот препарат и ту дозу, которые Вам назначил лечащий врач.
- Срок годности на этикетке предварительно заполненного шприца (ГОДЕН ДО:).
Не использовать шприц, если истек последний день указанного месяца. - Описание препарата Дарбэстим®. Раствор должен быть прозрачным или бесцветным или. Если раствор мутный или содержит частицы, препарат использовать нельзя.
- Вы можете заметить маленькие пузырьки воздуха в предварительно заполненном шприце. Вам не нужно удалять пузырьки воздуха перед инъекцией. Введение раствора с пузырьками воздуха является безопасным.
- Тщательно вымыть руки.
- Выбрать комфортное, хорошо освещенное место и чистую поверхность, удобно расположить все необходимые материалы.
Как выбрать место инъекции?
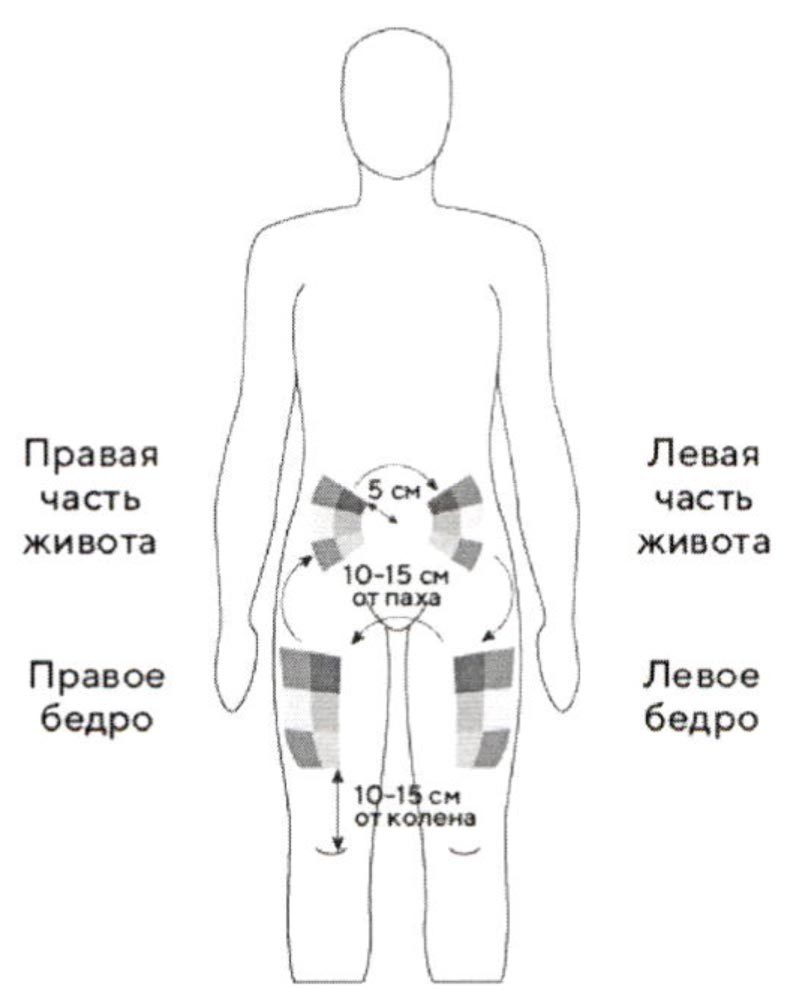
Лучше всего делать инъекции в верхнюю часть бедер и живота. Если инъекции Вам делает кто-то другой, можно использовать тыльную поверхность рук.
Если область, куда Вы собрались делать инъекцию, покраснела или отекла, следует выбрать другое место инъекции.
Раздел 2. Рекомендации по введению препарата Дарбэстим®
Как подготовиться к инъекции?
Перед инъекцией Вы должны сделать следующее:
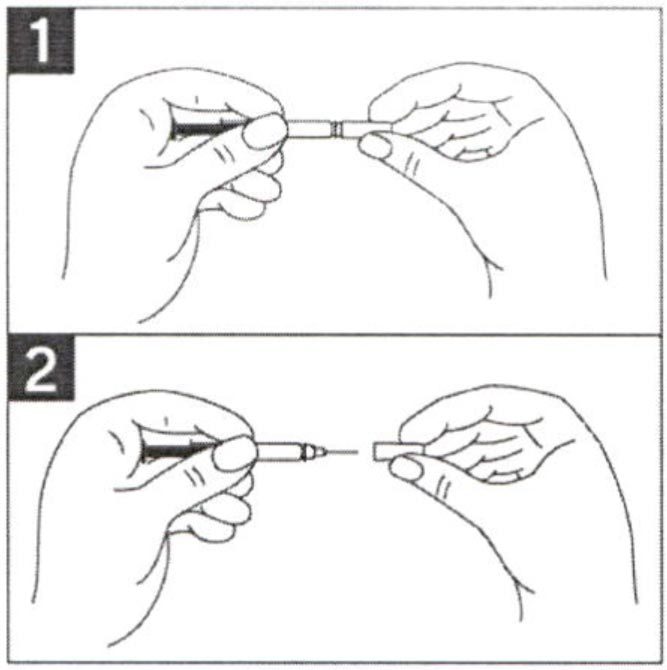
- Во избежание изгиба иглы, осторожно потянуть колпачок с иглы сразу, без скручивания, как показано на рисунках 1 и 2.
- Не дотрагиваться до иглы и не нажимать на поршень.
- Теперь Вы можете использовать предварительно заполненный шприц.
Как вводить препарат?
- Продезинфицировать место инъекции с помощью спиртовой салфетки, и зажать кожу (не сдавливая) большим и указательным пальцами.
- Ввести иглу в кожу полностью так, как показывал врач или медицинская сестра.
- Ввести назначенную дозу подкожно, как показывал Ваш врач или медицинская сестра.
- Медленно и непрерывно надавливать на поршень, при этом сжимать кожную складку и не отпускать ее, пока шприц не опустеет.
- Извлечь иглу и отпустить складку кожи.
- Если выступит кровь, аккуратно вытереть ее спиртовой салфеткой. Не растирать место инъекции. При необходимости, можно заклеить его пластырем.
- Один предварительно заполненный шприц предназначен для однократного применения. Не использовать оставшийся в шприце препарат Дарбэстим®.
Помните: При возникновении сложностей, обратитесь за помощью или советом к лечащему врачу или медицинской сестре.
В случае введения препарат Дарбэстим® в предварительно заполненном шприце с защитным устройством для иглы, Вам надо знать, что каждый шприц оснащен предохранителем иглы, который активируется автоматически для закрытия иглы после завершения инъекции.
Как вводить препарат Дарбэстим® в предварительно заполненном шприце с защитным устройством для иглы?
- Очистить кожу с помощью спиртовой салфетки.
- Во избежание загиба иглы, острожное потянуть колпачок с иглы сразу. Не дотрагиваться до иглы и не нажимать на поршень.
- Зажать кожу (не сдавливая) большим и указательным пальцами. Ввести иглу в кожу.
- Надавливать на поршень с легким непрерывным давлением. Надавливать на поршень следует до тех пор, пока шприц не опустеет. Прозрачный предохранитель иглы не сработает, пока предварительно заполненный шприц не опустеет. Пока поршень нажат, удалите иглу из кожи, отпустите поршень и позвольте шприцу подняться вверх до тех пор, пока вся игла не покроется прозрачным защитным устройством для иглы. Если защитное устройство для иглы не активизировалось, возможно, что Вы не выполнили инъекцию полностью. Позвоните Вашему лечащему врачу, если Вы считаете, что Вы не получили полную дозу.
- Если выступит кровь, аккуратно вытереть ее спиртовой салфеткой. Не растирать место инъекции. При необходимости, можно заклеить его пластырем.
Побочное действие
Общие положения
Выявленные нежелательные реакции, связанные с приемом дарбэпоэтина альфа, включают гипертензию, инсульт, тромбоэмболии, судороги, аллергические реакции, сыпь/эритему и парциальную красноклеточную аплазию (ПККА).
Боль в месте инъекции была зарегистрирована, как связанная с лечением, в исследованиях, в которых дарбэпоэтин альфа вводили в виде подкожных инъекций. Дискомфорт в месте инъекции в целом был слабым и преходящим и развивался преимущественно после первой инъекции.
Частота развития нежелательных реакций указана по классу системы органов и частоте возникновения.
Частота возникновения определена следующим образом: очень часто ≥1/10); часто (≥1/100, <1/10); нечасто (≥1/1000, <1/100); редко (≥1/10000, <1/1000); очень редко (<1/10000), неизвестно (не может быть оценена по имеющимся данным). Данные представлены отдельно для пациентов с ХПН и онкологических пациентов и отражают профиль нежелательных реакций в этих популяциях.
Пациенты с хронической почечной недостаточностью
В контролируемых исследованиях из 1357 пациентов, 766 пациентов получали дарбэпоэтин альфа и 591 пациент получал рекомбинантный эритропоэтин человека. 83% находились на диализе, 17% – нет. Инсульт был выявлен в качестве нежелательной реакции в дополнительном клиническом исследовании (TREAT).
Частота нежелательных реакций, расцененных как связанные с лечением дарбэпоэтином альфа, составляет:
| Система органов по MedDRA | Частота возникновения | Нежелательная реакция |
| Со стороны крови и лимфатической системы | Неизвестна* | Парциальная красноклеточная аплазия |
| Со стороны иммунной системы | Очень часто* | Гиперчувствительность |
| Со стороны нервной системы | Часто | Инсульт |
| Нечасто* | Судороги | |
| Со стороны сердца | Очень часто | Повышение артериального давления |
| Со стороны сосудов | Редко | Тромбоэмболии |
| Со стороны кожи и подкожной клетчатки | Часто | Сыпь/Эритема |
| Со стороны организма в целом, включая местные реакции | Часто | Боль в месте инъекции |
*см. описание отмеченных реакций.
Онкологические больные
Нежелательные реакции были определены на основании объединенных данных семи рандомизированных двойных слепых плацебо-контролируемых исследований дарбэпоэтина альфа, включавших 2112 пациентов (дарбэпоэтин альфа 1200, плацебо 912). В клинические исследования включались пациенты с солидными опухолями (например, легких, молочной железы, толстой кишки, яичников) и лимфоидными злокачественными новообразованиями (например, лимфомой, множественной миеломой). Частота нежелательных эффектов, расцененных как связанных с лечением дарбэпоэтином альфа, составляет:
| Система органов по MedDRA | Частота возникновения | Нежелательная реакция |
| Со стороны иммунной системы | Очень часто* | Гиперчувствительность |
| Со стороны нервной системы | Нечасто* | Судороги |
| Со стороны сердца | Часто* | Гипертензия |
| Со стороны сосудов | Часто | Тромбоэмболии, включая тромбоэмболию легочной артерии |
| Со стороны кожи и подкожной клетчатки | Часто | Сыпь/Эритема |
| Со стороны организма в целом, включая местные реакции | Очень часто | Отек |
| Часто | Боль в месте инъекции |
*см. описание отмеченных нежелательных реакций
Описание отмеченных нежелательных реакций
Пациенты с хронической почечной недостаточностью
Инсульт сообщался как распространенная нежелательная реакция у пациентов с ХПН в исследовании TREAT.
В отдельных случаях сообщалось о нейтрализующих антителах к эритропоэтину, опосредующих парциальную красноклеточную аплазию (ПККА), связанную с терапией дарбэпоэтином альфа, преимущественно у пациентов с ХПН, получавших препарат подкожно. В случае подтверждения диагноза ПККА, терапия дарбэпоэтином альфа должна быть прекращена, и пациенты не должны быть переведены на другой рекомбинантный эритропоэтин.
Частота реакций гиперчувствительности оценивалась на основе данных клинических исследований как «очень часто» у пациентов с ХПН. Сообщалось о развитии серьезных реакций гиперчувствительности, включая анафилактические реакции, ангионевротический отек, аллергический бронхоспазм, кожную сыпь и крапивницу, связанные с приемом дарбэпоэтина альфа.
Сообщалось о судорогах у пациентов, получающих дарбэпоэтин альфа. Частота оценивалась на основе данных клинических исследований как «нечасто» у пациентов с ХНП.
Онкологические пациенты
При применении в рутинной клинической практике, у онкологических пациентов наблюдалась гипертензия. Частота оценивалась на основе данных клинических исследований как «часто» у онкологических пациентов и также часто встречалась в группах, получающих плацебо.
При применении в рутинной клинической практике, у онкологических пациентов наблюдались реакции гиперчувствительности. Частота реакций гиперчувствительности оценивалась на основе данных клинических исследований как «очень часто» у онкологических пациентов. Также реакции гиперчувствительности очень часто встречались в группах, получающих плацебо. Сообщалось о развитии серьезных реакций гиперчувствительности, включающих анафилактические реакции, ангионевротический отек, аллергический бронхоспазм, кожную сыпь и крапивницу, связанных с приемом дарбэпоэтина альфа.
При применении в рутинной клинической практике у пациентов, получающих дарбэпоэтин альфа, сообщалось о судорогах. Частота оценивалась на основе данных клинических исследований как «нечасто» у онкологических пациентов. Судороги часто встречались в группах, получающих плацебо.
Дети с хронической почечной недостаточностью
Имеются ограниченные данные по безопасности применения дарбэпоэтина альфа у детей. Безопасность дарбэпоэтина альфа оценивалась в клиническом исследовании у детей с ХПН (от 1 года до 18 лег), получающих диализ, которые были стабильны при приеме эпоэтина альфа и затем, для поддержания уровня гемоглобина, были переведены на дарбэпоэтин альфа. Не было выявлено дополнительных нежелательных реакций у детей, по сравнению с ранее зарегистрированными у взрослых пациентов.
Передозировка
Максимальное количество дарбэпоэтина альфа, безопасное для введения в виде однократной или многократной дозы, не определялось. В случае недостаточного контроля уровня гемоглобина и титрования дозы, терапия дарбэпоэтином альфа может привести к полицитемии. После передозировки дарбэпоэтином альфа наблюдались случаи тяжелой гипертензии.
В случае выявления полицитемии введение дарбэпоэтина альфа следует временно прекратить (см. раздел «Способ применения и дозы»). При наличии клинических показаний может быть выполнена флеботомия.
Взаимодействие с другими лекарственными средствами
Клинические данные, полученные до настоящего времени, не содержат указаний на взаимодействие дарбэпоэтина альфа с другими веществами. Однако известно, что потенциально возможно его взаимодействие с препаратами, характеризующимися высокой степенью сродства к эритроцитам, такими как циклоспорин, такролимус. При одновременном назначении дарбэпоэтина альфа с подобными лекарственными средствами, следует контролировать уровень их содержания в сыворотке крови с модификацией дозы в случае повышения концентрации гемоглобина.
Ввиду того, что исследования по совместимости не проводились, дарбэпоэтин альфа не следует смешивать или вводить в виде инфузии вместе с другими медицинскими препаратами.
Особые указания
Общие положения
Необходим мониторинг артериального давления у всех пациентов, особенно в начале терапии препаратом Дарбэстим®. При не достижении адекватного контроля артериального давления стандартными методами, концентрация гемоглобина может быть снижена путем уменьшения дозы и отмены препарата Дарбэстим® (см. раздел «Способ применения и дозы»).
При лечении препаратом Дарбэстим® пациентов с ХПН, отмечалось развитие тяжелой формы гипертензии, включая гипертонический криз, гипертоническую энцефалопатию и судорожные припадки.
С целью подтверждения эффективности эритропоэза, всем больным следует определять содержание железа до и во время лечения с целью назначения, в случае необходимости, дополнительной терапии препаратами железа.
Отсутствие ответа па применение препарата Дарбэстим® должно служить стимулом для выявления причинных факторов. Эффективность эритропоэз-стимулирующих препаратов (ЭСП) снижается при недостатке в организме железа, фолиевой кислоты или витамина В12, вследствие чего уровень их содержания необходимо корректировать. Эритропоэтический ответ также может быть ослаблен при наличии сопутствующих инфекционных заболеваний, симптомов воспаления или случаев травмы, скрытой кровопотери, гемолиза, тяжелой алюминиевой интоксикации, сопутствующих гематологических заболеваний или фиброза костного мозга. Численность ретикулоцитов следует рассматривать как один из параметров оценки. Если типичные причины отсутствия ответа исключены, а у больного выявляется ретикулоцитопения, следует провести исследование костного мозга. Если картина костного мозга соответствует картине ПККА, рекомендуется выполнить исследование на присутствие антител к эритропоэтину.
Была описана ПККА, вызванная нейтрализующим действием антиэритропоэтиновых антител, связанная с применением ЭСП, включая и дарбэпоэтин альфа. Чаще всего такие сообщения касались пациентов с ХПН, получавших препарат подкожно. Было показано, что эти антитела перекрестно реагируют со всеми эритропоэтическими белками. В случае постановки диагноза ПККА, лечение препаратом дарбэпоэтина альфа необходимо прекратить без последующего перевода на терапевтический режим, включающий другой рекомбинантный эритропоэтический белок (см. раздел «Побочное действие»).
Парадоксальное снижение уровня гемоглобина и развитие тяжелой анемии с низким уровнем ретикулоцитов должно приводить к немедленной отмене лечения эпоэтином и проведению теста на наличие антител к эритропоэтину. Такие случаи были описаны у пациентов с гепатитом С, которые получали терапию интерфероном и рибавирином в сочетании с эпоэтинами. Применение эпоэтинов в лечении анемии при гепатите С не одобрено.
Во всех исследованиях дарбэпоэтина альфа критерием исключения были активные заболевания печени, поэтому данные о применении препарата у больных с нарушением функции печени отсутствуют. Так как печень считается основным путем выведения рчЭпо, пациентам с патологией печени эти препараты следует назначать с осторожностью.
Злоупотребление дарбэпоэтином альфа у здоровых лиц может привести к избыточному повышению гематокрита. Подобные явления могут быть ассоциированы с опасными для жизни осложнениями со стороны сердечно-сосудистой системы.
При поддержании уровня гемоглобина у пациентов с хронической почечной недостаточностью, его концентрация не должна превышать верхней границы, указанной в разделе «Способ применения и дозы». В ходе клинических исследований, при достижении целевого уровня гемоглобина более 120 г/л на фоне применения ЭСП, у пациентов отмечался повышенный риск смертности, развития серьезных осложнений со стороны сердечно-сосудистой системы или нарушений мозгового кровообращения, включая инсульт и тромбоз сосудистого доступа.
В ходе контролируемых клинических исследований не удалось выявить значительных преимуществ от применения эпоэтинов, если концентрация гемоглобина превышает уровень, необходимый для контроля симптомов анемии и устранения потребности в гемотрансфузиях.
Дарбэстим® следует применять с осторожностью у пациентов с эпилепсией. Имеются сообщения о возникновении судорог у пациентов, получавших дарбэпоэтин альфа.
Больные с хронической почечной недостаточностью
У пациентов с хронической почечной недостаточностью, поддерживающие концентрации гемоглобина не должны превышать верхнюю границу целевой концентрации гемоглобина, рекомендованной в разделе «Способ применения и дозы». В ходе клинических исследований наблюдались повышенные риски летального исхода, серьезных сердечно-сосудистых осложнений или нарушений мозгового кровообращения, включая инсульт, и тромбоз сосудистого доступа при назначении ЭСП для достижения уровня гемоглобина свыше 120 г/л (7,5 ммоль/л).
Проводимые контролируемые клинические исследования не показали значительных преимуществ при назначении эпоэтинов, когда концентрация гемоглобина повышается выше уровня, необходимого для лечения симптомов анемии и для того, чтобы избежать переливания крови.
Всем пациентам с уровнем ферритина сыворотки крови ниже 100 мкг/л или тем, у кого насыщение трансферрина ниже 20%, рекомендуется дополнительное лечение препаратами железа.
Во время применения препарата Дарбэстим® следует регулярно контролировать сывороточное содержание калия. Повышение концентрации калия было описано у нескольких пациентов, получающих дарбэпоэтин альфа, однако причинная связь установлена не была. При выявлении повышенной или повышающейся концентрации калия, введение дарбэпоэтина альфа следует прекратить до ее нормализации.
Онкологические больные
Влияние на рост опухоли
Эпоэтины представляют собой факторы роста, которые, главным образом, стимулируют выработку эритроцитов. Рецепторы к эритропоэтину могут экспрессироваться па поверхности различных опухолевых клеток. Как и в случае любых факторов роста, существует предположение о том, что эритропоэтины способны стимулировать рост опухолей.
В ряде контролируемых клинических исследований у онкологических больных, получающих химиотерапию, применение эпоэтинов не увеличивало общую продолжительности жизни или не снижало риск прогрессии опухоли у пациентов, с анемией, ассоциированной с онкологическим заболеванием.
В контролируемых клинических исследованиях дарбэпоэтина альфа и других ЭСП было продемонстрировано:
- Уменьшение времени до прогрессии у пациентов с распространенным раком головы и шеи, получающими лучевую терапию, при корректирующем назначении эпоэтина до достижения целевого значения гемоглобина выше, чем 140 г/л, ЭСП не показаны данной группе пациентов.
- Уменьшение общей продолжительности жизни и повышение смертности, связанное с прогрессией заболевания за 4 месяца у пациентов с метастазирующим раком молочной железы, получавших химиотерапию, при корректирующем назначении эпоэтина до достижения, целевого значения гемоглобина 120-140 г/л.
- Повышение риска смерти при корректирующем назначении эпоэтина до достижения целевого значения гемоглобина 120 г/л у пациентов с активной злокачественной опухолью, не получавших ни химиотерапии, ни лучевой терапии. Эритропоэз-стимулирующие агенты не показаны данной группе пациентов.
В соответствии с вышеизложенным, в некоторых клинических ситуациях для лечения анемии у пациентов с онкологическими заболеваниями следует применять переливание крови. Решение о назначении рекомбинантных эритропоэтинов следует принимать на основании оценки соотношения польза/риск для каждого индивидуального пациента, принимая во внимание особенности клинической ситуации. Необходимо учитывать следующие факторы: вид и стадия опухолевого процесса; степень анемии; ожидаемая продолжительность жизни; обстановка, в которой пациент будет проходить лечение; и пожелания самого пациента (см. раздел «Фармакодинамика»).
У больных с солидными опухолями или с лимфопролиферативными злокачественными заболеваниями при возрастании уровня содержания гемоглобина выше 120 г/л следует, строго соблюдать схему адаптации дозы, описанную в разделе «Способ применения и дозы», с целью минимизации потенциального риска развития тромбоэмболических явлений. Также необходимо регулярно контролировать численность тромбоцитов и концентрацию гемоглобина в крови.
Особые указания по использованию
Дарбэпоэтин альфа представляет собой стерильный продукт, изготовленный без консервантов.
Одним шприцом следует вводить не более одной дозы препарата. Любое количество лекарственного препарата, оставшееся в предварительно заполненном шприце, подлежит уничтожению.
Перед введением раствор препарата Дарбэстим® следует проконтролировать на предмет присутствия видимых частиц. Допускается использование только бесцветного, прозрачного или слабо опалесцирующего раствора. Раствор нельзя встряхивать. Перед введением следует дождаться прогревания ПЗШ до комнатной температуры.
Чтобы избежать возникновения дискомфорта в месте инъекции, необходимо менять места введения препарата.
Любые количества неиспользованного препарата или его отходов подлежат уничтожению в соответствии с местными требованиями.
Влияние на способность управлять транспортными средствами и механизмами
Влияние дарбэпоэтина альфа на способность к вождению автомобиля и обращению с техникой выявлено не было.
Форма выпуска
Раствор для инъекций.
Первый вариант упаковки:
Предварительно заполненные шприцы по 0,4 мл раствора (25 мкг/мл) – 10 мкг, или 0,375 мл раствора (40 мкг/мл) – 15 мкг, или 0,5 мл раствора (40 мкг/мл) – 20 мкг или 0,3 мл раствора (100 мкг/мл) – 30 мкг, или 0,4 мл раствора (100 мкг/мл) – 40 мкг, или 0,5 мл раствора (100 мкг/мл) – 50 мкг, или 0,3 мл раствора (200 мкг/мл) – 60 мкг, или 0,4 мл раствора (200 мкг/мл) – 80 мкг, или 0,5 мл раствора (200 мкг/мл) – 100 мкг, или 0,3 мл раствора (500 мкг/мл) – 150 мкг или 0,6 мл раствора (500 мкг/мл) – 300 мкг, или 1,0 мл раствора (500 мкг/мл) – 500 мкг в шприцы из стекла 1 гидролитического класса.
На каждый шприц наклеивают этикетку.
По 1 шприцу помещают в контурную ячейковую упаковку из пленки ПВХ. По 1 или 4 контурные ячейковые упаковки (по 0,3; 0,375; 0,4 или 0,5 мл препарата) или по 1 контурной ячейковой упаковке (по 0,6 или 1,0 мл препарата) помещают вместе с инструкцией по применению в пачку из картона. Пачка со шприцами дополнительно комплектуется спиртовыми салфетками в количестве 1 или 4 шт.
Второй вариант упаковки:
Предварительно заполненные шприцы по 0,4 мл раствора (25 мкг/мл) – 10 мкг, или 0,375 мл раствора (40 мкг/мл) – 15 мкг, или 0,5 мл раствора (40 мкг/мл) – 20 мкг или 0,3 мл раствора (100 мкг/мл) – 30 мкг, или 0,4 мл раствора (100 мкг/мл) – 40 мкг, или 0,5 мл раствора (100 мкг/мл) – 50 мкг, или 0,3 мл раствора (200 мкг/мл) – 60 мкг, или 0,4 мл раствора (200 мкг/мл) – 80 мкг, или 0,5 мл раствора (200 мкг/мл) – 100 мкг, или 0,3 мл раствора (500 мкг/мл) – 150 мкг или 0,6 мл раствора (500 мкг/мл) – 300 мкг, или 1,0 мл раствора (500 мкг/мл) – 500 мкг в шприцы из стекла 1 гидролитического класса.
На каждый шприц наклеивают этикетку.
По 1 шприцу с системой защиты иглы помещают в контурную ячейковую упаковку из пленки ПВХ. По 1 или 4 контурные ячейковые упаковки (по 0,3; 0,375; 0,4 или 0,5 мл препарата) или но 1 контурной ячейковой упаковке (по 0,6 или 1,0 мл препарата) помещают вместе с инструкцией по применению в пачку из картона. Пачка со шприцами дополнительно комплектуется спиртовыми салфетками в количестве 1 или 4 шт.
Условия хранения
Хранить при температуре 2-8°С в защищенном от света месте. Не замораживать.
Хранить в недоступном для детей месте.
Срок годности
3 года. Не использовать по окончанию срока годности.
Условия отпуска из аптек
Отпускают по рецепту.
Владелец регистрационного удостоверения
ЗАО «БИОКАД», Россия, 198515, г. Санкт-Петербург, Петродворцовый район, п. Стрельна, ул. Связи, д. 34, литер А.
Производитель
ЗАО «БИОКАД», Россия, 143422, Московская обл., Красногорский р-н, с. Петрово-Дальнее.
Организация, принимающая претензии потребителей
ЗАО «БИОКАД», 198515, г. Санкт-Петербург, Петродворцовый район, п. Стрельна, ул. Связи, д. 34, лит. А.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Инструкция по применению Аранесп
- Состав
- Показания к применению
- Противопоказания к применению
- Рекомендации по применению
- Применение при беременности и кормлении грудью
- Фармакологическое действие
- Побочные действия
- Особые указания
- Передозировка
- Лекарственное взаимодействие
- Условия хранения
- Условия отпуска
Состав
Активное вещество: Дарбэпоэтин альфа (рекомбинантный) 500 мкг (500 мкг/мл).
Вспомогательные вещества : натрия дигидрофосфата моногидрат — 2,118 мг, натрия гидрофосфат — 0,661 мг, натрия хлорид — 8,182 мг, полисорбат 80 — 0,05 мг, вода для инъекций — до 1 мл.
Показания к применению Аранесп
- Лечение анемии, ассоциированной с хронической почечной недостаточностью, у взрослых и детей 11 лет и старше;
- лечение симптоматической анемии у взрослых онкологических больных с немиелоидными злокачественными новообразованиями, получающих химиотерапию.
Противопоказания к применению Аранесп
- Повышенная чувствительность к дарбэпоэтину альфа, рчЭпо или к любому компоненту препарата.
- Плохо контролируемая артериальная гипертензия.
С осторожностью
- Заболевания печени;
- серповидно-клеточная анемия;
- эпилепсия.
Рекомендации по применению
Лечение препаратом Аранесп должно проводиться врачами, имеющими опыт назначения по вышеупомянутым показаниям.
Аранесп (SureClick) поставляется готовым для применения в предварительно заполненных шприц-ручках (ПЗШР). ПЗШР предназначаются только для подкожных инъекций. Инструкции по применению препарата, обращению с ним и порядку его уничтожения приводятся в разделе Особые указания по использованию.
Лечение анемии у больных с хронической почечной недостаточностью.
Аранесп можно вводить подкожно или внутривенно. Подкожное введение предпочтительно для больных, не получающих гемодиализ, для предотвращения пункции периферических вен. Целью терапии является повышение содержания гемоглобина до уровня, превышающего 110 г/л. Для каждого пациента необходим индивидуальный подбор требуемого гемоглобина выше 110 г/л. Следует избегать повышения гемоглобина более чем на 20 г/л за 4 недели или гемоглобина выше 140 г/л. Клинические исследования показали, что индивидуальный ответ пациента может быть различным. Тем не менее, на начальных этапах следует придерживаться приведенных ниже рекомендаций, как для взрослых, так и для детей, с последующей оптимизацией терапии в зависимости от клинических показаний.
Лечение препаратом Аранесп включает две стадии- фаза коррекции и поддерживающая фаза:
Фаза коррекции.
Начальная доза при подкожном или внутривенном введении должна составлять 0,45 мкг/кг массы тела при однократном еженедельном введении. Альтернативно, для больных, не получающих диализ, допускается подкожное введение препарата в начальной дозе 0,75 мкг/кг массы тела каждые две недели. Если повышение концентрации гемоглобина оказывается недостаточным (менее 10 г/л за 4 недели), доза препарата увеличивается приблизительно на 25%. Повышение дозы препарата не должно осуществляться чаще, чем один раз в четыре недели.
Если концентрация гемоглобина возрастает более чем на 25 г/л за четыре недели, дозу Аранеспа следует снизить на 25-50% в зависимости от скорости повышения гемоглобина. Если содержание гемоглобина превышает 140 г/л терапию следует прервать до снижения гемоглобина ниже 130 г/л, а затем возобновить введение препарата в дозе, сниженной примерно на 25% относительно предыдущей. Гемоглобин следует измерять еженедельно или раз в две недели до его стабилизации. Впоследствии гемоглобин можно оценивать периодически.
Поддерживающая фаза.
В период поддерживающей фазы можно продолжать однократное еженедельное введение препарата Аранесп или перейти на введение каждые две недели. При переводе пациентов, находящихся на диализе, с еженедельных инъекций на режим введения однократно раз в две недели, исходная доза должна вдвое превышать дозу, вводившуюся один раз в неделю. Для пациентов, не получающих диализа, после достижения требуемой концентрации гемоглобина на фоне назначения препарата раз в две недели, его подкожное введение может производиться один раз в месяц с использованием исходной дозы вдвое превышающей предыдущую дозу, вводившуюся раз в две недели.
Титрование дозы с целью поддержания требуемой концентрации гемоглобина следует производить так часто, как это требуется.
Для каждого пациента необходим индивидуальный подбор требуемого гемоглобина выше 110 г/л. Если для поддержания требуемого гемоглобина необходима оптимизация дозы Аранеспа, ее рекомендуется увеличивать приблизительно на 25%. В случае, если наблюдается повышение гемоглобина более, чем 20 г/л за 4 недели, дозу препарата следует уменьшить приблизительно на 25%, в зависимости от скорости повышения, Если содержание гемоглобина превышает 140 г/л, терапию следует прервать до уменьшения концентрации ниже 130 г/л, а затем возобновить введение препарата в дозе, меньше примерно на 25% относительно предыдущей.
После любого изменения дозы или режима введения, содержание гемоглобина следует контролировать каждую 1 или 2 недели. Изменение дозы во время поддерживающей фазы должно выполняться не чаще одного раза в две недели.
При изменении пути введения препарата следует использовать те же дозы препарата и осуществлять мониторинг концентрации гемоглобина раз в 1-2 недели с целью поддержания требуемого уровня гемоглобина.
Пациентов, получающих еженедельно по одной, две или три инъекции рчЭпо, можно перевести на режим однократного еженедельного введения Аранеспа или его введение один раз в две недели. Исходную еженедельную дозу Аранеспа (мкг/неделю) определяют,разделив общую еженедельную дозу рчЭпо (МЕ/неделю) на 200. Исходную дозу Аранеспа (мкг/ в две недели) при режиме введения один раз в две недели определяют путем деления суммарной кумулятивной дозы рчЭпо, введенного за двухнедельный период, на 200. Ввиду известной индивидуальной вариабельности, для отдельных больных может потребоваться титрование доз до получения оптимального терапевтического эффекта.
При замещении рчЭпо на препарат Аранесп. измерение уровня гемоглобина следует выполнять не реже одного раза в неделю или в две недели, а способ введения препарата должен оставаться неизменным.
Лечение симптоматической анемии у онкологических больных.
Аранесп следует вводить подкожно пациентам с анемией (при концентрации гемоглобина ≤110 г/л.)
Рекомендованная начальная доза составляет 500 мкг (6,75 мкг/кг) при введении раз в три недели. Если клинический ответ через девять недель неадекватен (утомляемость, содержание гемоглобина), дальнейшая терапия может оказаться неэффективной.
Альтернативно, один раз в неделю препарат можно вводить в дозе 2,25 мкг/кг массы тела.
Применение Аранеспа прекращают примерно через четыре недели после завершения химиотерапии.
Содержание гемоглобина не должно превышать 130 г/л.
После достижения целевого гемоглобина дозу препарата следует снизить на 25-50% для поддержания гемоглобина на соответствующем уровне. При необходимости допускается дальнейшее снижение дозы, чтобы не допустить возрастания концентрации гемоглобина более 130 г/л.
Если скорость возрастания гемоглобина превышает 20 г/л в течение 4 недель, дозу препарата следует снизить на 25-50%.
Применение Аранесп при беременности и кормлении грудью
Адекватных и строго контролируемых клинических исследований по безопасности применения Аранесп при беременности не проводилось. С осторожностью и после тщательной оценки ожидаемой пользы терапии для матери и потенциального риска для плода следует назначать препарат беременным женщинам. При необходимости назначения Аранеспа в период лактации грудное вскармливание следует прекратить.
Фармакологическое действие
Фармакодинамика
Дарбэпоэтин альфа стимулирует эритропоэз по тому же механизму, что и эндогенный эритропоэтин. Дарбэпоэтин альфа содержит пять N-связанных углеводных цепей, в то время как эндогенный гормон я рекомбинантные человеческие эритропоэтины (рчЭпо) имеют всего три цепи. Дополнительные остатки Сахаров, с молекулярной точки зрения, не отличаются от таковых, представленных в эндогенном гормоне. Вследствие повышенного содержания углеводов дарбэпоэтин альфа обладает более длительным периодом полувыведения в сравнении с рчЭпо, а следовательно, и большей активностью in vivo.
Несмотря на указанные изменения молекулярной структуры дарбэпоэтин альфа сохраняет очень узкую специфичность к эритропоэтиновому рецептору.
Доклинические данные по безопасности.
Во всех исследованиях на крысах и собаках при применении Аранеспа значимо возрастала концентрация гемоглобина, гематокрита, эритроцитов и ретикулоцитов, что соответствует ожидаемому фармакологическому эффекту. Нежелательные явления при введении очень высоких доз препарата рассматривались как следствие усиленного фармакологического действия (снижения тканевого кровотока вследствие увеличения вязкости крови). Сюда же были отнесены миелофиброзы и гипертрофия селезенки, а также расширение комплекса QRS на ЭКГ у собак, без нарушения cердечного ритма и влияния на интервал QT. Аранесп не обладал каким-либо генотоксическим потенциалом и не оказывал влияния на пролиферацию клеток негематологического ряда ни in vitro, ни in vivo. В исследованиях по хронической токсичности не наблюдалось туморогенного или неожиданного митогенного ответа ни в одном изученном типе тканей. В продолжительных исследованиях на животных оценка канцерогенного потенциала дарбэпоэтина альфа не выполнялась.
В испытаниях, проводившихся на крысах и кроликах, не наблюдалось клинически значимого влияния на беременность, эмбриональное/фетальное развитие, роды или постнатальное развитие. Уровень проникновения препарата через плаценту был минимальным. Изменений фертильности не отмечалось.
Фармакокинетика
В связи с повышенным содержанием углеводов концентрация циркулирующего в крови дарбэпоэтина альфа превышает минимальную концентрацию необходимую для стимуляции эритропоэза в течение более продолжительного времени в сравнении с эквивалентными дозами рчЭпо, что позволяет снизить частоту введения дарбэпоэтина альфа с сохранением эквивалентного уровня биологического ответа.
Больные с хронической почечной недостаточностью.
Фармакокинетика дарбэпоэтина альфа была изучена у больных с хронической почечной недостаточностью при внутривенном и подкожном введении препарата. Его период полувыведения составлял 21 час (стандартное отклонение (СО) 7,5) при внутривенном введении. Клиренс дарбэпоэтина альфа составил 1,9 мл/час/кг (СО 0,56), а объем распределения (Орс) был приблизительно эквивалентен объему плазмы (50 мл/кг). При подкожном введении препарата его биодоступность соответствовала 37%. При ежемесячном подкожном введении дарбэпоэтина альфа в дозе от 0,6 до 2,1 мкг/кг его период полувыведения составлял 73 часа (СО 24). Более продолжительный период полувыведения дарбопоэтина альфа при подкожном введении, по сравнению с внутривенным, обусловлен кинетикой абсорбции. В ходе клинических исследований минимальное накопление препарата наблюдалось при любом способе введения. В доклинических исследованиях было продемонстрировано, что почечный клиренс дарбэпоэтина минимален (до 2% общего клиренса) и не оказывает влияния на период полувыведения препарата из сыворотки.
Способ введения не влияет на дозу дарбэпоэтина альфа, необходимую для поддержания достигнутого гемоглобина.
Онкологические больные, получающие химиотерапию.
После подкожного введения препарата в дозе 2,25 мкг/кг взрослым онкологическим больным средние максимальные концентрации дарбэпоэтина альфа, составляющие 10,6 нг/мл (СО 5,9), устанавливались в среднем в течение 91 часа (СО 19,7). Эти параметры соответствовали линейной фармакокинетике дозы в широком диапазоне значений (от 0,5 до 8 мкг/кг при еженедельном введении и от 3 до 9 мкг/кг при введении раз в две недели). Фармакокинетические параметры не изменялись при многократном дозировании в течение 12 недель (еженедельное введение или введение раз в две недели). Отмечалось ожидаемое умеренное повышение (менее 2-кратного) сывороточной концентрации препарата при достижении равновесного состояния, но не было выявлено признаков его накопления при повторном назначении. ФК исследования были выполнены с привлечением пациентов с индуцированной во время химиотерапии анемией, которые в комбинации с химиотерапией подкожно получали инъекции дарбэпоэтина альфа в дозе 6,75 мкг/кг раз в три недели. В данном исследовании среднее значение (СО) периода полувыведения составляло 74 (СО 27) часа.
Побочные действия Аранесп
В связи с применением дарбэпоэтина альфа были описаны редкие случаи развития потенциально значимых серьезных аллергических реакций, включавших диспноэ, кожные высыпания и крапивницу.
Больные с хронической почечной недостаточностью.
К нежелательным эффектам, развивающимся при лечении Аранеспом, относятся артериальная гипертензия и тромбоз сосудистого доступа. Однако, в объединенной базе данных по безопасности ни одно их подобных явлений не было ассоциировано с изменением гемоглобина (менее 120 против >120 г/л) или со скоростью повышения уровня гемоглобина (менее 10, от 10 до менее 20, от 20 до менее 30 и > 30 г/л гемоглобина в течение 4 недель).
Боль в месте подкожной инъекции регистрировалась чаще, чем при применении рчЭпо.
Дискомфорт в месте инъекции, как правило, был легким и временным и развивался, главным образом, после первой инъекции.
В очень редких случаях у пациентов с хронической почечной недостаточностью, получающих Аранесп, наблюдались судороги. В отдельных случаях, в связи с применением Аранеспа была описана парциальная красноклеточная аплазия (ПККА), вызванная нейтрализующим влиянием анти-эритропоэтиновых антител.
Все остальные связанные с лечением нежелательные явления наблюдались с 1% частотой и реже (не часто или редко встречающиеся). Большинство из них имели легкую или умеренную степень выраженности и соответствовали сопутствующим заболеваниям у подобных больных.
Онкологические больные.
Частота развития артериальной гипертензии и сердечно-сосудистых явлений была сравнима у онкологических больных, получавших плацебо, рчЭпо или Аранесп, когда Аранесп вводился подкожно. Кроме того, подобные нежелательные явления не были ассоциированы ни с содержанием гемоглобина (менее 130 против >130 г/л), ни со скоростью повышения уровня гемоглобина (> 20 г/л в течение четырех недель). У онкологических больных, получавших Аранесп, повышалась частота тромбоэмболических осложнений, включая тромбоз глубоких вен и эмболию легочной артерии, по сравнению с пациентами, получавшими плацебо.
В целом, нежелательные явления при применении Аранеспа у онкологических больных, получавших сопутствующую химиотерапию, соответствовали основному заболеванию и применяемой для его лечения химиотерапии.
Наиболее часто описываемым нежелательным явлением, сочтенным связанным с применением Аранеспа (менее 5%), была боль в месте инъекции. Дискомфорт в месте инъекции был, как правило, легким и временным.
Особые указания
С целью подтверждения эффективности эритропоэза всем больным следует определять содержание железа до и во время лечения с целью назначения, в случае необходимости, дополнительной терапии препаратами железа.
Отсутствие ответа на применение Аранеспа должно служить стимулом для выявления причинных факторов. Эффективность стимулирующих эритропоэз веществ снижается при недостатке в организме железа, фолиевой кислоты или витамина В12, вследствие чего уровень их содержания необходимо корректировать. Эритропоэтический ответ также может быть ослаблен при наличии сопутствующих инфекционных заболеваний, симптомов воспаления или случаев травмы, скрытой кровопотери, гемолиза, тяжелой алюминиевой интоксикации, сопутствующих гематологических заболеваний или фиброза костного мозга. Численность ретикулоцитов следует рассматривать как один из параметров оценки. Если типичные причины отсутствия ответа исключены, а у больного выявляется ретикулоцитопения, следует провести исследование костного мозга. Если картина костного мозга соответствуют картине ПККА, рекомендуется выполнить исследование на присутствие антител к эритропоэтину.
ПККА, вызванная нейтрализующим действием анти-эритропоэтиновых антител, была описана в связи с применением рекомбинантных эритропоэтических белков, включая и дарбэпоэтин альфа. Было показано, что эти антитела перекрестно реагируют со всеми эритропоэтическими белками. В случае постановки диагноза ПККА, лечение препаратом Аранесп необходимо прекратить без последующего перевода пациента на терапевтический режим, включающий другой рекомбинантный эритропоэтический белок (см. раздел Побочное действие).
Во всех исследованиях Аранеспа критерием исключения были активные заболевания печени, таким образом, данные о применении препарата у больных с нарушением функции печени отсутствуют. Так как печень считается основным путем выведения Аранеспа и рчЭпо, пациентам с патологией печени эти препараты следует назначать с осторожностью.
Злоупотребление Аранеспом у здоровых лиц может привести к избыточному повышению гематокрита, Подобные явления могут быть ассоциированы с опасными для жизни осложнениями со стороны сердечно-сосудистой системы.
Защитный колпачок иглы на ПЗШР содержит в своем составе натуральную обезвоженную резину (производное латекса), что может явиться причиной аллергической реакции.
Больные с хронической почечной недостаточностью.
Использование дополнительной терапии препаратами железа рекомендуется всем пациентам, у которых концентрация ферритина в сыворотке не превышает 100 мкг/л или уровень насыщения трансферрина ниже 20%, Артериальное давление необходимо контролировать у всех больных, в особенности в начале применения Аранеспа. Больные должны быть осведомлены относительно важности соблюдения рекомендаций по применению антигипертензивных препаратов и диетических ограничений. Если артериальное давление плохо поддается контролю при проведении соответствующих процедур, можно снизить содержание гемоглобина за счет уменьшения дозы Аранеспа или временного прекращения его введения (см. раздел Способ применения и дозы).
У больных с хронической почечной недостаточностью и клиническими симптомами ишемической болезни сердца или застойной сердечной недостаточности, целевые уровни гемоглобина следует определять индивидуально. У таких больных максимальное содержание гемоглобина не должно превышать 120 г/л, за исключением случаев, когда тяжесть симптомов (например, стенокардия) требует другого решения.
Во время применения Аранеспа следует регулярно контролировать сывороточное содержание калия. Повышение концентрации калия было описано у нескольких пациентов, получающих Аранесп, однако причинная связь установлена не была. При выявлении повышенной или повышающейся концентрации калия, введении Аранеспа следует прекратить до ее нормализации.
Пациентам, страдающим эпилепсией, следует назначать Аранесп с осторожностью, так как имеются сообщения о развитии судорог у больных с хронической почечной недостаточностью, получавших Аранесп.
Онкологические больные
Влияние на рост опухоли.
Эритропоэтины представляют собой факторы роста, которые, главным образом, стимулируют выработку эритроцитов. Рецепторы к эритропоэтину могут экспрессироваться на поверхности различных опухолевых клеток. Как и в случае любых факторов роста, существует предположение о том, что эритропоэтины способны стимулировать рост злокачественных новообразований любого типа. В двух контролируемых клинических исследованиях по применению эритропоэтинов у больных с разными формами рака, включая рак головы и шеи и рак молочной железы, было продемонстрировано необъясненное повышение уровня смертности.
У больных с солидными опухолями или с лимфопролиферативными злокачественными заболеваниями при возрастании уровня содержания гемоглобина выше 130 г/л следует строго соблюдать схему адаптации дозы, описанную в разделе Способ применения и дозы, с целью минимизации потенциального риска развития тромбоэмболических явлений. Также необходимо регулярно контролировать численность тромбоцитов и концентрацию гемоглобина в крови.
Особые указания по использованию
Аранесп представляет собой стерильный продукт, изготовленный без консервантов. Одной шприц-ручкой следует вводить не более одной дозы препарата. Любое количество лекарственного препарата, оставшееся в предварительно заполненной шприц ручке, подлежит уничтожению.
Перед введением раствор препарата Аранесп следует проконтролировать на предмет присутствия видимых частиц. Допускается использование только бесцветного, прозрачного или слабо опалесцирующего раствора. Раствор нельзя встряхивать. Перед введением следует дождаться прогревания ПЗШР до комнатной температуры.
Чтобы избежать возникновения дискомфорта в месте инъекции, необходимо менять места введения препарата.
Любые количества неиспользованного продукта или его отходов подлежат уничтожению в соответствии с местными требованиями.
Передозировка
Границы терапевтической дозы препарата Аранесп очень широкие. Даже при очень высокой концентрации препарата в сыворотке крови, не наблюдалось симптомов передозировки.
В случае выявления полицитемии введение Аранеспа следует временно прекратить. При наличии клинических показаний может быть выполнена флеботомия.
Лекарственное взаимодействие
Клинические данные, полученные до настоящего времени, не содержат указаний на взаимодействие Аранеспа с другими веществами. Однако известно, что потенциально возможно его взаимодействие с препаратами, характеризующимися высокой степенью сродства к эритроцитам, такими как циклоспорин, такролимус. При одновременном назначении дарбэпоэтина альфа с любыми подобными лекарственными средствами, следует контролировать уровень их содержания в сыворотке крови с модификацией дозы в случае повышения концентрации гемоглобина.
Ввиду того, что исследования по совместимости не проводились, препарат Аранесп не следует смешивать или вводить в виде инфузии вместе с другими медицинскими препаратами.
Условия хранения
Хранить в защищенном от света месте, при температуре от 2 °С до 8 °С.
Срок годности — 2 года.
Условия отпуска
Отпускается по рецепту
Состав
В 1 мл раствора для подкожного и внутривенного введения содержится 500 МЕ или 2000 МЕ эпоэтина бета.
Форма выпуска
1 мл раствора в стеклянной ампуле; 5 ампул в контурной упаковке; 1 или 2 упаковки в пачке из бумаги.
Фармакологическое действие
Стимулирующее действие на гемопоэз.
Фармакодинамика и фармакокинетика
Фармакодинамика
Эритропоэтин – это очищенный гликопротеин, рекомбинантный аналог природного Эритропоэтина человека, который представляет собой фактор роста гематопоэза. Произведен благодаря генно-инженерным методам.
Увеличивает количество ретикулоцитов, эритроцитов, активирует синтез гемоглобина в эритроцитах. Не влияет на лейкопоэз. Считается, что препарат взаимодействует с эритропоэтиновыми рецепторами на стенках клеток.
Фармакокинетика
После введения подкожно наибольшая концентрация в крови регистрируется через 14-26 часов. Время полувыведения составляет 15-26 часов , при внутривенном введении – до 12 часов.
Показания к применению
- Для увеличения количества крови для последующей аутотрансфузии, чтобы избежать гемотрансфузии донорской крови.
- Тяжелая анемия на фоне почечной недостаточности хронического типа.
- Анемия при болезнях костного мозга и ряде хронических заболеваний (СПИД, миелодиспластических заболеваниях, онкологических болезнях).
- Предупреждение анемии у недоношенных детей (до 34-й недели) с массой тела при рождении 0,75-1,5 кг.
Противопоказания
Артериальная гипертензия (не поддающаяся контролю), инфаркт миокарда или инсульт в предшествующем лечению месяце, тромбоэмболия в прошлом, нестабильная стенокардия, сенсибилизация к эпоэтину бета.
Побочные действия
- Явления со стороны нервной системы: энцефалопатия, боль в голове, спутанность сознания.
- Явления со стороны системы кровообращения: гипертонический криз, артериальная гипертония.
- Аллергические реакции: сыпь, реакции анафилактоидного типа.
- Явления со стороны свертывания крови и действие на анализ крови: тромботические осложнения, тромбоцитоз.
- Явления со стороны метаболизма: гиперкалиемия.
Инструкция по применению (Способ и дозировка)
Инструкция на Эритропоэтин советует разрабатывать схему лечения индивидуально в зависимости от степени анемии, состояния больного и вида болезни.
Возможно внутривенное и подкожное введение. Первоначальная доза составляет 50-150 МЕ на килограмм веса, рекомендуемая кратность введения – через день.
Передозировка
При передозировке могут возникать эффекты, характерные для высокой степени активности действующего вещества. При очень высоком содержании гемоглобина возможно произведение флеботомии. Применяется симптоматическое лечение.
Взаимодействие
Препараты, активирующие кроветворение (препараты железа и другие) могут усиливать действие эпоэтина бета на гемопоэз.
Условия продажи
Возможно приобрести только по рецепту.
Условия хранения
- Хранить в темном месте.
- Хранить в диапазоне температур 2-8 градусов.
- Беречь от детей.
- Не замораживать.
Срок годности
Три года.
Особые указания
Советуется с осторожностью использовать средство при тромбоцитозе, эпилепсии, печеночной или сосудистой недостаточности, при злокачественных онкологических заболеваниях, у пациентов с нефросклерозом.
Эффективность терапии уменьшается при недостатке железа в организме, а также при инфекционно-воспалительных поражениях или гемолизе.
Во время лечения советуется воздерживаться от управления мобильными механизмами и автотранспортом.
Аналоги Эритропоэтина
Совпадения по коду АТХ 4-го уровня:
Другие препараты Эритропоэтина (аналоги): Веро-Эпоэтин, Эпотал, Рекормон, Эпрекс, Эпостим, Эпоэтин, Эритростим, Эритростим, Шанпоэтин, Гемакс, Бинокрит, Вепокс.
При беременности и лактации
В данные периоды возможен прием препаратов эпоэтина бета при наличии строгих показаний.
Отзывы
Эритропоэтин и его аналоги являются препаратами выбора при анемиях тяжелой степени. Существует мало средств с таким выраженным действием на гемопоэз. Побочные явления при использовании средства встречаются нечасто.
Цена Эритропоэтина, где купить
Купить Эритропоэтин в Москве 2000 МЕ №5 обойдется в в среднем в 2850 рублей. Цена Эритропоэтина на Украине в несколько раз дороже, так как там можно встретить только аналоги препарата.
Эфлейра 60мг/мл 1мл 2 шт. раствор для подкожного введения
Эфлейра
Инструкция по применению Эфлейра 60мг/мл 1мл 2 шт. раствор для подкожного введения
Состав и форма выпуска
Раствор — 1 мл.:
- Действующее вещество: нетакимаб – 60 мг;
- Вспомогательные вещества: натрия ацетата тригидрат – 1,74 мг, трегалозы дигидрат – 80 мг, полоксамер 188 – 0,5 мг, уксусная кислота ледяная – до pH 5,0, вода для инъекций – до 1,0 мл.
Раствор для подкожного введения 60 мг/мл.
Преднаполненные шприцы
По 1,0 мл препарата в трехкомпонентные шприцы. Цилиндр каждого шприца изготовлен из бесцветного нейтрального стекла I гидролитического класса. Шприц с одной стороны имеет впаянную иглу для инъекций из нержавеющей стали, которая защищена жестким колпачком. С другой стороны шприц укупорен поршнем, шток которого изготовлен из полипропилена; на конце поршня имеется уплотнитель из бутилкучука, ламинированный фторполимером.
На каждый шприц наклеивают самоклеящуюся этикетку.
По 1 шприцу помещают в контурную ячейковую упаковку из пленки ПВХ или ПЭТ.
По 2 контурных ячейковых упаковки вместе с инструкцией по применению помещают в пачку из картона. Пачку дополнительно комплектуют спиртовыми салфетками в количестве 2 шт.
Автоинжектор
По 1,0 мл препарата в трехкомпонентные шприцы. Цилиндр каждого шприца изготовлен из бесцветного нейтрального стекла I гидролитического класса.
Шприц с препаратом встроен в пластиковый автоинжектор, состоящий из защитного колпачка, корпуса с прозрачной зоной контроля, верхнего корпуса и кнопки активации. Внутреннее устройство автоинжектора имеет цилиндр, защищающий иглу, и поршень, который через систему механического пуска соединен с кнопкой активации.
На каждый автоинжектор наклеивают самоклеящуюся этикетку.
По 1 автоинжектору помещают в контурную ячейковую упаковку из пленки ПВХ или ПЭТ.
По 2 контурных ячейковых упаковки вместе с инструкцией по применению помещают в пачку из картона. Пачку дополнительно комплектуют спиртовыми салфетками в количестве 2 шт.
Регистрационный номер:
Торговое наименование: Эфлейра®
Международное непатентованное или группировочное наименование: нетакимаб
Лекарственная форма: раствор для подкожного введения
Состав
1 мл раствора содержит:
Действующее вещество: нетакимаб – 60 мг;
Вспомогательные вещества: натрия ацетата тригидрат – 1,74 мг, трегалозы дигидрат – 80 мг, полоксамер 188 – 0,5 мг, уксусная кислота ледяная – до pH 5,0, вода для инъекций – до 1,0 мл.
Описание
Прозрачный или слегка опалесцирующий от бесцветного до светло-желтого или светло-желтого с коричневатым оттенком цвета раствор.
Фармакотерапевтическая группа
Интерлейкина ингибитор
Код по АТХ: L04AC
Цена на Эфлейра в Москве
Купить Эфлейра
Фармакологические свойства
Фармакодинамика
Нетакимаб является рекомбинантным гуманизированным моноклональным антителом, в терапевтических концентрациях специфически связывающим интерлейкин-17А (ИЛ-17А), находящийся непосредственно в тканях или в крови и других биологических жидкостях. ИЛ-17А – провоспалительный цитокин, гиперпродукция которого преимущественно обусловлена активацией Th17-лимфоцитов. В рамках врожденного иммунитета ИЛ-17А выполняет защитную роль. При хронических иммуновоспалительных заболеваниях патологическая активация Th17-лимфоцитов и гиперпродукция ИЛ-17 стимулирует Т-клеточный ответ и усиленную продукцию других медиаторов воспаления: ИЛ-1, ИЛ-6, фактора некроза опухоли альфа (ФНО-α), факторов роста (Г-КСФ, ГМ-КСФ) и различных хемокинов.
Нетакимаб обладает высокой термодинамической константой специфического связывания с ИЛ-17А человека. По данным доклинических исследований специфическое связывание нетакимаба в нормальных тканях человека ограничено тканями легкого, тимуса, лимфатического узла, миндалин, что согласуется с данными об экспрессии ИЛ-17 клетками этих тканей.
Применение нетакимаба не сопровождается статистически значимым изменением уровня Т-лимфоцитов и не влияет на уровень и соотношение иммуноглобулинов классов А, G и М.
Специфическая противовоспалительная активность нетакимаба продемонстрирована в тестах in vitro
и in vivo. Нетакимаб дозозависимо ингибирует ИЛ-17 и ФНОα-зависимую продукцию интерлейкина-6 на культуре клеток при IC50 40 pM. На модели коллаген-индуцированного артрита у яванских макак (Macaca fascicularis) многократное (один раз в неделю в течение 4-х недель) подкожное введение нетакимаба сопровождается снижением выраженности воспалительной реакции в суставах, что подтверждено при гистологическом исследовании (суставной хрящ остается интактным, синовиальные оболочки — без признаков поражения и воспалительной реакции, пролиферации синовиоцитов не отмечено).
У больных псориазом использование нетакимаба сопровождается угасанием явлений воспаления и гиперкератоза в коже, достоверным снижением уровня С-реактивного белка и СОЭ. У пациентов с активным анкилозирующим спондилитом и псориатическим артритом на фоне применения нетакимаба отмечается уменьшение симптомов воспаления в позвоночнике, энтезисах и суставах, а также быстрое снижение концентрации
С-реактивного белка, являющегося маркером воспаления.
Фармакокинетика
Всасывание/распределение
Изменение концентрации нетакимаба после его подкожного введения является дозозависимым (значения Сmax, Cmax-mult, AUC0-t находятся в прямой зависимости от дозы). Препарат характеризуется медленной фазой абсорбции с постепенным линейным нарастанием концентрации в сыворотке крови.
При однократном подкожном введении нетакимаба в дозе 120 мг пациентам с бляшечным псориазом препарат начинал обнаруживаться в сыворотке крови в течение 0,5 — 4 часов после введения; максимальная концентрация нетакимаба (Сmax) составляла 15,1 [7,7-19,3] мкг/мл, время ее достижения – 144 [72-168] ч, AUC0-168 – 1667,8 [932,2-2270,8] (мкг/мл)·ч.
При повторных введениях отмечено накопление препарата в сыворотке крови с ростом концентрации в 1,8-3,6 раза. Максимальная концентрация при многократном введении (Cmax-mult) составляла 33,0 [23,1-44,0] мкг/мл и достигалась (Tmax-mult) через 1680 [672-2016] часов.
Выведение
Характеристики выведения нетакимаба являются типичными для препаратов на основе моноклональных антител: показатели Kel, T1/2, MRT, Cl не зависят от дозы вводимого препарата, период полувыведения после однократного введения составляет около 16 суток.
Средний клиренс нетакимаба при однократном введении в дозе 120 мг пациентам с бляшечным псориазом составил 1,8 л/сутки.
Фармакокинетические параметры нетакимаба у пациентов с анкилозирующим спондилитом схожи с таковыми у пациентов с бляшечным псориазом.
Пациенты с почечной и печеночной недостаточностью: фармакокинетические данные у больных с почечной и печеночной недостаточностью отсутствуют.
Пациенты в возрасте старше 65 лет:
фармакокинетические данные у лиц в возрасте старше 65 лет отсутствуют.
Показания к применению
· Лечение бляшечного псориаза среднетяжелой и тяжелой степени у взрослых пациентов, когда показана системная терапия или фототерапия.
· Лечение активного анкилозирующего спондилита у взрослых пациентов при недостаточном ответе на стандартную терапию.
· Лечение активного псориатического артрита в режиме монотерапии или в комбинации с метотрексатом при недостаточном ответе на стандартную терапию.
Противопоказания
· Гиперчувствительность к нетакимабу, а также к любому из вспомогательных веществ препарата.
· Клинически значимые инфекционные заболевания в острой фазе, включая туберкулез.
· Детский и подростковый возраст до 18 лет.
· Беременность, грудное вскармливание.
С осторожностью
Следует соблюдать осторожность при назначении нетакимаба пациентам с хроническими и рецидивирующими инфекциями или с анамнестическими указаниями на них, в периоде ранней реконвалесценции после тяжелых и среднетяжелых инфекционных заболеваний, а также после недавно проведенной вакцинации живыми вакцинами.
В связи с ограниченными данными клинических исследований о применении нетакимаба у пациентов в возрасте старше 65 лет, следует соблюдать осторожность при назначении препарата пациентам указанной возрастной группы.
В связи с отсутствием сведений о применении нетакимаба у больных воспалительными заболеваниями кишечника, следует избегать его назначения пациентам с болезнью Крона или язвенным колитом.
Применение во время беременности и в период грудного вскармливания
Применение при беременности
При применении нетакимаба у животных не выявлено отрицательного влияния на репродуктивную функцию, эмбриотоксичности или тератогенных эффектов. Исследований влияния на плод у беременных женщин не проводилось. Препарат противопоказан к применению во время беременности.
Применение при грудном вскармливании
Нет данных о проникновении нетакимаба в грудное молоко. Учитывая, что иммуноглобулины класса G, к которым относится нетакимаб, при циркуляции в крови матери могут выделяться с грудным молоком, применение в период грудного вскармливания противопоказано. Во избежание негативного воздействия на ребенка следует прекратить либо грудное вскармливание, либо терапию, учитывая соотношение риска и пользы для матери и ребенка.
Влияние на фертильность
В исследованиях у животных не обнаружено негативного воздействия нетакимаба на фертильность. Данные о влиянии препарата на фертильность у людей отсутствуют.
Способ применения и дозы
Применение препарата Эфлейра® должно осуществляться под наблюдением врачей, имеющих опыт лечения заболеваний, при которых показан препарат Эфлейра®. После соответствующего обучения возможно самостоятельное введение препарата пациентом при условии динамического наблюдения со стороны лечащего врача. Препарат Эфлейра® может применяться как в стационарных, так и в амбулаторных условиях.
Препарат Эфлейра®
вводится в дозе 120 мг в виде двух подкожных инъекций по 1 мл препарата с концентрацией 60 мг/мл.
Лечение бляшечного псориаза среднетяжелой и тяжелой степени у взрослых пациентов, когда показана системная терапия или фототерапия: рекомендуемая доза 120 мг в виде двух подкожных инъекций по 1 мл (60 мг) препарата каждая вводится 1 раз в неделю на неделях 0, 1 и 2, затем 1 раз каждые 4 недели.
Лечение активного анкилозирующего спондилита при недостаточном ответе на стандартную терапию: рекомендуемая доза 120 мг в виде двух подкожных инъекций по 1 мл (60 мг) препарата каждая. Препарат вводится 1 раз в неделю на неделях 0, 1 и 2, затем каждые 2 недели.
Лечение активного псориатического артрита в режиме монотерапии или в комбинации с метотрексатом при недостаточном ответе на стандартную терапию: рекомендуемая доза 120 мг в виде двух подкожных инъекций по 1 мл (60 мг) препарата каждая. Препарат вводится 1 раз в неделю на неделях 0, 1 и 2, затем каждые 2 недели до недели 10 включительно. Далее с недели 14 препарат вводится в дозе 120 мг в виде двух подкожных инъекций по 1 мл (60 мг) каждая 1 раз в 4 недели.
|
Показание |
Разовая доза |
Индукция |
Поддерживающая терапия |
|
Бляшечный псориаз |
120 мг |
0, 1, 2 недели |
1 раз в 4 недели, начиная с недели 6 |
|
Анкилозирующий спондилит |
120 мг |
0, 1, 2 недели |
1 раз в 2 недели, начиная с недели 4 |
|
Псориатический артрит |
120 мг |
0, 1, 2 недели |
1 раз в 2 недели, начиная с недели 4 по неделю 10 включительно, далее 1 раз в 4 недели с недели 14 |
При пропуске очередного введения по любой причине инъекция препарата Эфлейра®
должна быть произведена как можно быстрее. Дата следующего введения рассчитывается исходя из продолжительности задержки с введением препарата: если со времени пропуска введения прошло не более 3 дней, то следующую инъекцию препарата необходимо выполнить по текущему графику, если с момента пропуска введения прошло более 3 дней, то новый отсчет для даты следующего введения начинают с момента фактически проведенной инъекции препарата Эфлейра®.
Указания по применению
Подготовка к проведению подкожной инъекции
· Тщательно вымыть руки
· Достать упаковку с двумя шприцами/автоинжекторами с препаратом Эфлейра® из холодильника, затем извлечь из картонной пачки и ячейковой упаковки два шприца/автоинжектора и положить на чистую поверхность. Убедиться, что срок хранения препарата, указанный на картонной пачке и шприце/автоинжекторе, не истек.
Препарат Эфлейра® в преднаполненном шприце
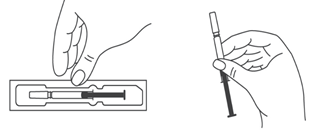
Препарат Эфлейра ® в автоинжекторе
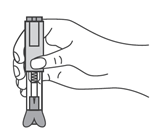
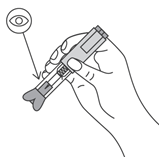
· Осмотреть шприцы/автоинжекторы, а также лекарственный препарат, находящийся в них. Препарат Эфлейра® представляет собой прозрачный раствор от бесцветного до светло-желтого цвета. Нельзя использовать препарат в случае:
— помутнения раствора, наличия в препарате посторонних видимых частиц;
— изменения цвета;
— повреждения любых частей шприца/автоинжектора;
— истечения срока годности (годен до…), указанного на картонной пачке, а также на этикетке шприца/автоинжектора.
· Перед введением шприцы/автоинжекторы с препаратом Эфлейра®
следует довести до комнатной температуры, для этого необходимо оставить шприц при комнатной температуре в течение приблизительно 25-30 минут. Не следует согревать шприц/автоинжектор каким-либо другим способом.
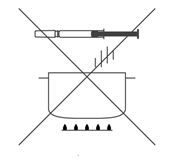
· Подготовить салфетку/ватный тампон, спиртовой раствор
На данном этапе не следует снимать колпачок шприца/автоинжектора
1. Техника выполнения подкожной инъекции препарата Эфлейра® в преднаполненном шприце
Выбор и подготовка места для инъекции
· Выбрать место инъекции (передняя брюшная стенка (отступая не менее 5 см от пупка), передняя и боковая поверхность бедра или средняя треть наружной части плеча (возможные места для инъекций закрашены на рисунке ниже));
· Места инъекций и стороны следует менять с каждой последующей процедурой инъекции. Расстояние между двумя введениями должно составлять как минимум 5 см.
· Нельзя вводить препарат в место на коже, где имеется болезненность, покраснение, уплотнение или кровоподтек (эти признаки могут указывать на наличие инфекции). По возможности не следует вводить препарат в псориатическую бляшку.
· Место для укола необходимо обработать спиртовой салфеткой/ватным тампоном круговыми движениями.
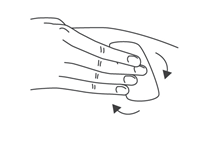
· Шприц не встряхивать.
· Снять колпачок с иглы, не дотрагиваясь до иглы и избегая ее прикосновения к другим поверхностям.
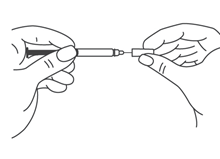
· Большим и указательным пальцами одной руки взять в складку обработанную кожу.
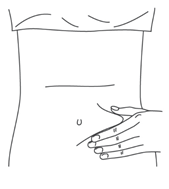
· В другую руку взять шприц, держа его градуированной поверхностью вверх. Введение препарата необходимо осуществлять под углом 45 или 90 градусов к поверхности кожи в зависимости от толщины кожи и выраженности подкожно-жирового слоя (у худощавых пациентов введение препарата осуществляется под углом 45 градусов, у пациентов с толщиной кожной складки более 1,5 см допустимо введение под углом 90 градусов).
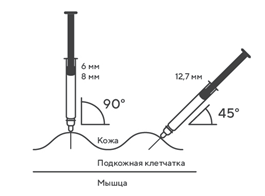
· Одним быстрым движением полностью ввести иглу в кожную складку.
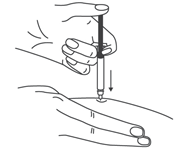
· После введения иглы складку кожи отпустить.
· Ввести весь раствор медленным постоянным надавливанием на поршень шприца в течение 2-5 секунд.
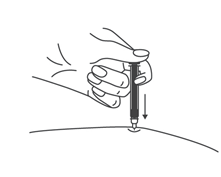
· Когда шприц будет пустым, вынуть иглу из кожи под тем же углом.
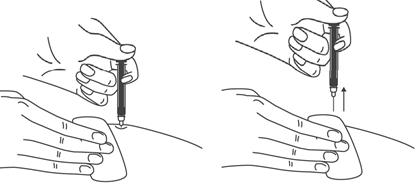
· Сухой стерильной салфеткой/ватным шариком слегка прижать область инъекции, но не растирать область выполненной инъекции. Из места инъекции может выделиться небольшое количество крови. При необходимости можно воспользоваться пластырем.
· Ввести вторую дозу препарата Эфлейра® аналогичным образом предпочтительно в ту же анатомическую область при условии, что место последующей инъекции должно быть не ближе 5 см от предыдущей. Время выполнения двух инъекций не должно превышать 10 минут.
После инъекции шприц повторно не использовать
2. Техника выполнения подкожной инъекции препарата Эфлейра® в автоинжекторе
· Устройство автоинжектора:
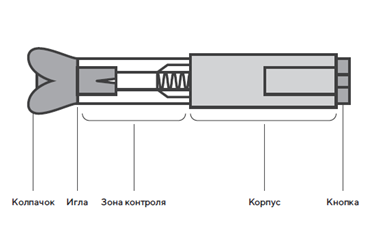
· Автоинжектор не встряхивать.
· Не снимайте защитный колпачок с автоинжектора до выполнения инъекции.
· Проверьте срок годности препарата по маркировке на упаковке, оцените целостность автоинжектора и состояние раствора через зону контроля автоинжектора. Не используйте автоинжектор в случае его повреждения, изменения цвета и прозрачности раствора, а также при истечении срока годности.
· Разместите автоинжектор на чистой горизонтальной поверхности.
· Выберите место введения (область брюшной стенки (отступая не менее 5 см от пупка) или передняя поверхность бедра) и обработайте кожу в области введения спиртовой салфеткой.
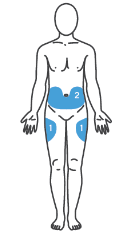
· Непосредственно перед инъекцией одной рукой снимите колпачок с автоинжектора, удерживая другой рукой его корпус.
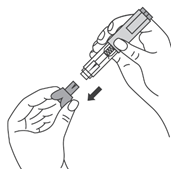
· Одной рукой возьмите в складку обработанную спиртовой салфеткой кожу и удерживайте в течение всей процедуры инъекции.
· В другую руку возьмите автоинжектор, удерживая его за корпус. Введение препарата необходимо осуществлять, плотно прижав автоинжектор под углом 90 градусов к поверхности кожи. Надавливание автоинжектором на поверхность кожной складки приведет к разблокированию кнопки.
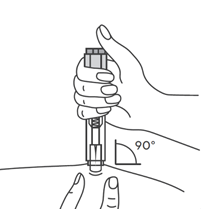
· Удерживая автоинжектор под углом 90 градусов к поверхности кожной складки, надавите до упора на кнопку автоинжектора. Вы услышите щелчок, указывающий на начало инъекции. Не изменяйте положение автоинжектора. Удерживайте кнопку в течение 10 секунд до окончания инъекции.
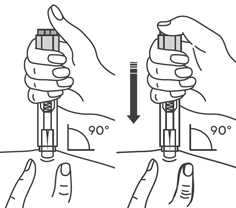
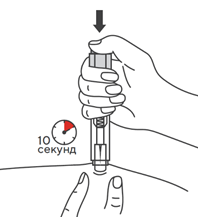
· Если инъекция не запустилась, отпустите кнопку и проверьте, плотно ли прижат автоинжектор к кожной складке. Прижмите плотно автоинжектор к коже и повторите процедуру инъекции, вновь нажав на кнопку.
· Удерживайте автоинжектор плотно прижатым к коже еще несколько секунд для полного введения лекарственного вещества. Осмотрев зону контроля, убедитесь, что вся доза препарата введена и контейнер пуст.
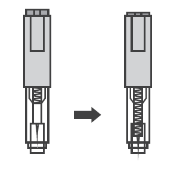
· После окончания инъекции извлеките автоинжектор, игла автоматически закроется защитным цилиндром.
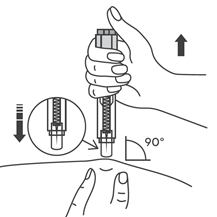
· Сухой стерильной салфеткой/ватным шариком слегка прижмите область инъекции, не растирая кожу. Из места инъекции может выделиться небольшое количество крови. При необходимости можно воспользоваться пластырем.
· Введите вторую дозу препарата Эфлейра®
аналогичным образом предпочтительно в ту же анатомическую область при условии, что место последующей инъекции должно быть не ближе 5 см от предыдущей. Время выполнения двух инъекций не должно превышать 10 минут.
После инъекции автоинжектор повторно не использовать
Утилизация расходного материала
Неиспользованный раствор препарата, использованные шприцы/автоинжекторы, салфетки/ватные тампоны и другие расходные материалы подлежат утилизации с применением закрывающегося, устойчивого к проколам контейнера для острых предметов из пластика или стекла. Не допускайте хранения использованных шприцев/автоинжекторов в местах, доступных для детей.
Побочное действие
В рамках проведенных клинических исследований у пациентов с бляшечным псориазом, псориатическим артритом и анкилозирующим спондилитом препарат Эфлейра® показал благоприятный профиль безопасности. Явлений дозолимитирующей токсичности не выявлено.
Большинство зарегистрированных нежелательных явлений (НЯ), связанных с применением препарата Эфлейра®, имели легкую или среднюю степень тяжести и не требовали прекращения терапии. Летальных исходов, связанных с терапией препаратом Эфлейра®, в ходе клинических исследований выявлено не было.
Наиболее частой нежелательной реакцией в проведенных клинических исследованиях была нейтропения, большинство случаев которой были легкой или средней степени тяжести, носили транзиторный характер и не требовали дополнительной терапии.
В данной инструкции нежелательные реакции представлены в соответствии с международным словарем нежелательных реакций MedDRA. Ниже приведен перечень нежелательных реакций, зарегистрированных у пациентов, получавших нетакимаб в рамках клинических исследований, и имеющих определенную, вероятную или возможную связь с приемом препарата. Частота указана с учетом следующих критериев: очень часто (≥1/10), часто (от ≥1/100 до <1/10), нечасто (от ≥1/1000 до <1/100), редко (от ≥1/10000 до <1/1000), очень редко (≤10000).
Инфекции и инвазии: часто – инфекции верхних дыхательных путей; нечасто – инфекция дыхательных путей, пневмония, назофарингит, фарингит, синусит, инфекция мочевыводящих путей, кандидоз пищевода, конъюнктивит вирусный, простой герпес, стафилококковое импетиго, фурункул, туберкулезная инфекция.
Нарушения со стороны печени и желчевыводящих путей: нечасто – гипербилирубинемия.
Желудочно-кишечные нарушения: нечасто – боль в животе, диарея.
Нарушения со стороны кожи и подкожной клетчатки: нечасто – экзема, дерматит, сыпь, зуд, крапивница.
Нарушения со стороны крови и лимфатической системы: часто – нейтропения, лейкопения, лимфоцитоз; нечасто – тромбоцитопения, лимфопения.
Нарушения со стороны иммунной системы: нечасто – гиперчувствительность.
Нарушения со стороны органа зрения:
нечасто – эписклерит.
Нарушения со стороны нервной системы: нечасто – головная боль, головокружение, парестезия, поражение лицевого нерва.
Нарушения со стороны сердца: нечасто — синусовая брадикардия, блокада левой ножки пучка Гиса.
Нарушения со стороны сосудов: нечасто — гипертензия (в том числе изолированная систолическая/диастолическая гипертензия), гипертонический криз.
Общие нарушения и реакции в месте введения: нечасто — гриппоподобное заболевание*, местная реакция**.
Нарушения метаболизма и питания: нечасто – гипергликемия.
Нарушения со стороны почек и мочевыводящих путей: нечасто – протеинурия.
Лабораторные и инструментальные данные: часто – повышение активности аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), положительный результат исследования на комплекс Mycobacterium tuberculosis; нечасто – повышение гамма-глутамилтрансферазы (ГГТ), повышение уровня холестерина в крови, увеличение веса.
Травмы, интоксикации и осложнения процедур: нечасто – головокружение во время процедуры (во время инъекции).
Доброкачественные, злокачественные и неуточненные новообразования (включая кисты и полипы):
нечасто – инфицированный невус.
Нарушения со стороны репродуктивной системы и молочных желез: нечасто – фиброзно-кистозная болезнь молочных желез.
*«Гриппоподобное заболевание» характеризуется появлением ряда симптомов, сходных с выявляемыми при гриппе или простуде, включая, например, но не ограничиваясь: повышение температуры, озноб, ломоту в теле, недомогание, слабость, снижение аппетита, сухой кашель, которые имеют временную связь с проведением инъекции препарата.
**Местные реакции могут включать в себя любые неблагоприятные проявления, возникающие в месте инъекции.
Передозировка
Клинические данные о передозировке отсутствуют. Максимальная переносимая доза для человека не установлена. В клинических исследованиях явлений дозолимитирующей токсичности не зарегистрировано: при подкожном введении нетакимаба в максимальной дозе 3 мг/кг (195 – 355 мг для взрослого человека с весом 65 – 85 кг) патологических отклонений не было выявлено.
Специфический антидот отсутствует. Лечение симптоматическое.
Взаимодействие с другими лекарственными препаратами
Сведений о наличии неблагоприятных лекарственных взаимодействий нетакимаба с другими лекарственными препаратами до настоящего времени не получено. Предполагается, что нетакимаб может усиливать иммуносупрессивное действие глюкокортикоидов, метотрексата, сульфасалазина, лефлуномида и других базисных противовоспалительных препаратов.
Смешивание препарата с другими лекарственными средствами, введение в виде инфузии строго запрещено.
Особые указания
Наличие таких потенциально тяжелых инфекций как ВИЧ, активный гепатит В и/или С, сифилис, туберкулез, относится к противопоказаниям для назначения нетакимаба. Применение нетакимаба у данной группы больных не изучено. Учитывая иммуносупрессивное действие нетакимаба, терапия этим препаратом потенциально может приводить к обострению хронических инфекций и повышению риска первичного инфицирования. Необходимо проводить скрининг и оценивать соотношение риска и пользы терапии нетакимабом у этих пациентов.
Пациентам с активным туберкулезом терапия препаратом Эфлейра® противопоказана. Перед назначением препарата Эфлейра® и в ходе терапии необходимо проводить стандартный скрининг на туберкулез. Пациентам с латентным туберкулезом рекомендуется пройти стандартный курс противотуберкулезной терапии перед началом терапии препаратом Эфлейра®.
При реактивации гепатита В терапия нетакимабом должна быть прекращена и назначена соответствующая противовирусная терапия.
Женщины детородного возраста и их половые партнеры должны использовать эффективные средства контрацепции в период проведения терапии.
При использовании нетакимаба потенциально возможно развитие реакции гиперчувствительности. В рамках проведенных клинических исследований нетакимаба анафилактических реакций не зарегистрировано, нечасто отмечались реакции гиперчувствительности и крапивница. Однако при использовании других ингибиторов ИЛ17 крапивница и анафилактические реакции отмечались в редких и очень редких случаях. При возникновении анафилактических или других серьёзных аллергических реакций применение препарата Эфлейра® следует немедленно прекратить и начать соответствующую симптоматическую терапию.
Наличие алкогольной или наркотической зависимости, а также психических расстройств может стать причиной несоблюдения пациентом графика лечения нетакимабом, что, в свою очередь может привести к снижению эффективности терапии. Необходим более тщательный мониторинг за пациентами с указанными состояниями в связи с отсутствием результатов клинических исследований на данной категории пациентов и возможностью повышенного риска развития гепатотоксичности и других неблагоприятных последствий.
Иммуногенность
В ходе клинических исследований препарата Эфлейра® при лечении псориаза, псориатического артрита и анкилозирующего спондилита выработка связывающих антител к нетакимабу была зарегистрирована менее чем в 0,5 % случаев. Нейтрализующих антител выявлено не было. Полученные данные демонстрируют низкую иммуногенность нетакимаба у пациентов с псориазом, псориатическим артритом и анкилозирующим спондилитом.
Пациенты в возрасте старше 65 лет
Данные об эффективности и безопасности препарата у пациентов в возрасте старше 65 лет ограничены. Не предполагается наличия существенных возрастных различий в распределении и выведении препарата.
Пациенты с нарушениями функции почек и печени
Эффективность и безопасность препарата у данной категории пациентов не изучались.
Пациенты моложе 18 лет, дети
Исследование эффективности и безопасности препарата у детей и лиц моложе 18 лет не проводилось.
Вакцинация
Не следует проводить иммунизацию живыми вакцинами в ходе лечения препаратом Эфлейра®, так как клиническая оценка безопасности данного взаимодействия в рамках клинических исследований не проводилась. Вакцинация живыми вакцинами до начала терапии препаратом Эфлейра®, а также интервал между вакцинацией и началом терапии должны соответствовать действующим клиническим рекомендациям. Иммунизация инактивированными вакцинами во время терапии нетакимабом должна выполняться с осторожностью.
Влияние на способность управлять транспортными средствами и механизмами
Отсутствуют данные о влиянии препарата Эфлейра® на способность управлять транспортными средствами и работать с машинами и (или) механизмами.
Форма выпуска
Раствор для подкожного введения 60 мг/мл.
Преднаполненные шприцы
По 1,0 мл препарата в трехкомпонентные шприцы. Цилиндр каждого шприца изготовлен из бесцветного нейтрального стекла I гидролитического класса. Шприц с одной стороны имеет впаянную иглу для инъекций из нержавеющей стали, которая защищена жестким колпачком. С другой стороны шприц укупорен поршнем, шток которого изготовлен из полипропилена; на конце поршня имеется уплотнитель из бутилкучука, ламинированный фторполимером.
На каждый шприц наклеивают самоклеящуюся этикетку.
По 1 шприцу помещают в контурную ячейковую упаковку из пленки ПВХ или ПЭТ.
По 2 контурных ячейковых упаковки вместе с инструкцией по применению помещают в пачку из картона. Пачку дополнительно комплектуют спиртовыми салфетками в количестве 2 шт.
Автоинжектор
По 1,0 мл препарата в трехкомпонентные шприцы. Цилиндр каждого шприца изготовлен из бесцветного нейтрального стекла I гидролитического класса.
Шприц с препаратом встроен в пластиковый автоинжектор, состоящий из защитного колпачка, корпуса с прозрачной зоной контроля, верхнего корпуса и кнопки активации. Внутреннее устройство автоинжектора имеет цилиндр, защищающий иглу, и поршень, который через систему механического пуска соединен с кнопкой активации.
На каждый автоинжектор наклеивают самоклеящуюся этикетку.
По 1 автоинжектору помещают в контурную ячейковую упаковку из пленки ПВХ или ПЭТ.
По 2 контурных ячейковых упаковки вместе с инструкцией по применению помещают в пачку из картона. Пачку дополнительно комплектуют спиртовыми салфетками в количестве 2 шт.
Условия хранения
При температуре от 2 до 8 °С в защищенном от света месте. Не замораживать!
Хранить в недоступном для детей месте.
Срок годности
2 года.
Не применять по истечении срока годности.
Условия отпуска
Отпускают по рецепту.
Купить Эфлейра в лучшей аптеке города по самой низкой цене.
Цены на Эфлейра в аптеках Москвы