Package insert / product label
Generic name: immunoglobulin g
Dosage form: subcutaneous injection
Drug class: Immune globulins
J Code (medical billing code): J1551 (100 mg, injection)
CUTAQUIG (Immune Globulin Subcutaneous (Human) — hipp)
16.5% solution
1. Indications and Usage for Cutaquig
CUTAQUIG (Immune Globulin Subcutaneous (Human) — hipp) is a 16.5% immune globulin solution for subcutaneous infusion (IGSC), indicated as replacement therapy for primary humoral immunodeficiency (PI) in adults and pediatric patients 2 years of age and older. This includes, but is not limited to, common variable immunodeficiency (CVID), X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.
2. Cutaquig Dosage and Administration
For subcutaneous use only
.
-
Before receiving treatment with CUTAQUIG: Obtain the patient’s serum Immunoglobulin G (IgG) trough level to guide subsequent dose adjustments (see
Dose Adjustment
).
2.1 Dose
-
Individualize the dose based on the patient’s pharmacokinetic and clinical response. Monitor serum IgG trough levels regularly to guide subsequent dose adjustments as needed (see
Dose Adjustment
). - Start CUTAQUIG treatment one week after the last Immune Globulin Intravenous (IGIV)/ Immune Globulin Subcutaneous (IGSC) infusion.
Dose for patients switching to CUTAQUIG from IGIV:
- Establish the initial weekly dose of CUTAQUIG by converting the monthly IGIV dose into an equivalent weekly dose and increasing it using a dose adjustment factor.
- To calculate the initial weekly dose of CUTAQUIG, divide the monthly IGIV dose in grams by the number of weeks between IGIV infusions and then multiply this value with a Dose Adjustment Factor of 1.30.
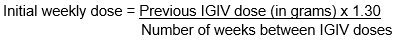
- To convert the dose (in grams) to milliliters (mL), multiply the calculated dose (in grams) by 6.
- Provided the total weekly dose is maintained, any dosing interval from daily up to weekly can be used and will result in systemic IgG exposure that is comparable to the previous IGIV treatment.
- For every other week dosing, multiply the calculated weekly CUTAQUIG dose by 2.
- For more frequent than weekly dosing (2-7 times a week) divide the calculated weekly CUTAQUIG dose by the desired number of infusions per week (e.g.: if you want to infuse CUTAQUIG 2 times per week, divide the weekly dose by 2).
- On average, serum IgG trough levels were approximately 23% higher during CUTAQUIG administration compared to those obtained during prior IGIV therapy.
-
To guide dose adjustments, see
Table 1
under
Dose Adjustment
.
Dose for patients switching to CUTAQUIG from IGSC:
- It is recommended to maintain the same weekly dosing (in grams) of CUTAQUIG that was used for the previous Immune Globulin Subcutaneous (Human) (IGSC) therapy (in grams).
- For every other week dosing, multiply the weekly dose by 2.
- For more frequent than weekly dosing (2-7 times a week) divide the weekly dose by the desired number of infusions per week (e.g.: if you want to infusion CUTAQUIG 2 times per week, divide the weekly dose by 2).
- To convert the dose (in grams) to milliliters (mL), multiply the calculated dose (in grams) by 6.
- Obtain a trough IgG level before switching, monitor clinical response and check the trough IgG level 2 to 3 months after initiating CUTAQUIG.
-
To guide dose adjustments, see
Table 1
under
Dose Adjustment
, if the trough IgG level during CUTAQUIG administration differs from the trough IgG level obtained during treatment with the previously administered IGSC product or target trough level.
Dose Adjustment:
Over time the dose may need to be individualized for each patient, depending on the pharmacokinetic and clinical response and on the desired IgG trough level. Measure the patient’s serum IgG trough level 2-3 months after switching to CUTAQUIG or after the last CUTAQUIG dose adjustment, in order to determine if a dose adjustment is necessary.
Calculate the difference between the patient’s target serum IgG trough level (in mg/dL) and the IgG trough level obtained during subcutaneous treatment with CUTAQUIG. Find this difference in column 1 of
Table 1
and according to frequency of administration (weekly or every other week) locate the corresponding adjustment amount of CUTAQUIG in mL/administration according to the body weight of the patient.
For more frequent dosing than weekly or every other week, add the weekly increment from
Table 1
to the weekly equivalent dose of the patient and then divide by the desired number of administrations per week.
Use the patient’s clinical response as the primary point to consider for any dose adjustment. Additional dose increments may be indicated based on the patient’s clinical response (i.e., infection frequency and severity).
Table 1 Incremental adjustment (mL) of weekly or every other week CUTAQUIG dosing based on the calculated difference between actual IgG trough level and the target trough level for the patient*
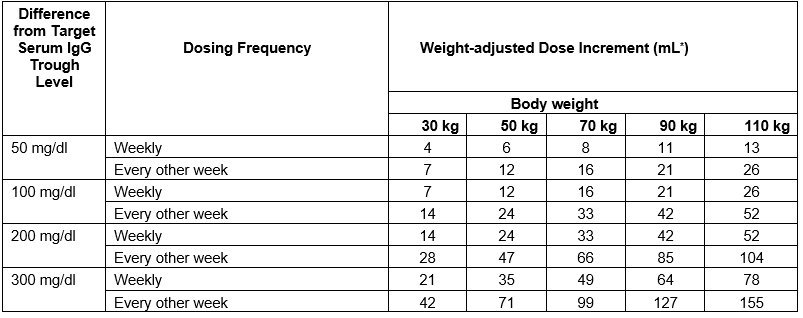
*
Derived from a linear regression model of trough levels and weekly dose per kg body weight.
For example: a patient with body weight of 70 kg is treated weekly and has a trough level of 600 mg/dL, but the target trough level is 900 mg/dL. The difference between the actual trough level (600) and the targeted trough level (900) is plus 300 mg/dL. Therefore, the recommended increase in the weekly dose would be approximately 49 mL.
A patient with a body weight of 50 kg, on an every other week dosing schedule and with an actual trough level of 900 mg/dL has a target trough level of 700 mg/dL. The difference between actual trough level (900) and targeted trough level (700) is minus 200 mg/dL, therefore the patient needs a decrease in his every other week dose of approximately 47 mL.
Monitor the patient’s clinical response and periodically check trough IgG levels, and repeat dose adjustment as needed.
Measles Exposure
If a patient has been exposed to measles, it may be prudent to administer a dose of Immune Globulin Intravenous as soon as possible and within 6 days of exposure. A dose of 400 mg/kg should provide a serum level > 240 mIU/mL of measles antibodies for at least two weeks.
If a patient is at risk of future measles exposure and receives a dose of less than 245 mg/kg subcutaneously per week, increase the dose to 245 mg/kg.
2.2 Preparation
CUTAQUIG is a clear and colorless solution that may turn to slightly opalescent and pale yellow during storage. Do not use the solution if it appears cloudy or contains particulates.
- Use aseptic technique when preparing and administering CUTAQUIG.
- Prior to administration visually inspect each vial of CUTAQUIG for particulate matter, whenever the solution and container permit.
- Do not mix CUTAQUIG with other products.
- Do not dilute CUTAQUIG.
- Do not shake the solution.
- Check the labeling for the expiration date and do not use beyond this date.
- CUTAQUIG vials are for single use only. Discard any unused product after each infusion in accordance with local requirements.
- Do not freeze. Do not use frozen product.
- Record lot number in patient’s medical history.
2.3 Administration
CUTAQUIG is for subcutaneous use only.
Administer CUTAQUIG by a healthcare provider, or by caregiver or self-administered by the patient after appropriate training.
Infuse CUTAQUIG in the following areas: abdomen, thigh, upper arm, and/or upper leg/hip area.
CUTAQUIG is intended for subcutaneous use using an infusion pump and compatible syringe(s).
Infuse CUTAQUIG simultaneously into up to 6 different sites. Infusion sites should be at least 2 inches (5 cm) apart. Rotate infusion sites between subsequent administrations.
Volume:
For patients not already on IGSC therapy, do not exceed the maximum initial volume per injection site of 25 mL/site for adults (≥17 years), 15 mL/site (for ages 7-16 years) and 10 mL/site (for ages 2-6 years) for the first 2 infusions.
For subsequent infusions, if tolerated, you may gradually increase infusion volume/site by approximately 10 mL per infusion site in adults (≥17 years) and by approximately 5-10 mL per infusion site in children (2-16 years) every 2-4 weeks.
The maximum infusion volume is 40 mL/site (for adults ≥17 years), 29 mL/site (for ages 7-16 years) and 15.5 mL/site (for ages 2-6 years).
Rate:
For patients not already on IGSC therapy, do not exceed the maximum recommended flow rates per hour per infusion site of 20 mL per hour per site for adults (≥17 years) and 15 mL per hour per site for children (2-16 years) for the first 2 infusions.
For subsequent infusions, if tolerated, you may gradually increase the infusion rate by approximately 10mL/hr/site in adults (≥17 years) and by approximately 5-10 mL/hr/site in children (2-16 years) every 2-4 weeks.
The maximum infusion rate is 52 mL per hour per site for adults (≥17 years) and 25 mL per hour per site for children (2-16 years).
Table 2 Infusion Volumes and Infusion Rates (Infusion rate and infusion volume per site may gradually be increased every 2-4 weeks as tolerated by patients)
Infusion parameters* |
Infusion Number |
||
First 2 infusions |
Subsequent infusions |
||
| Volume (mL/site) |
Adults ≥17 years |
≤ 25 | gradually increase by approximately 10 mL/site every 2-4 weeks up to a maximum of 40 |
| 7-16 years | ≤ 15 | gradually increase by approximately 5-10 mL/site every 2-4 weeks up to a maximum of 29 | |
| 2-6 years | ≤ 10 | gradually increase by approximately 5-10 mL/site every 2-4 weeks up to a maximum of 15.5 | |
| Rate (mL/hr/site) |
Adults ≥17 years |
≤ 20 | gradually increase by approximately 10 mL/hr/site every 2-4 weeks up to a maximum of 52 |
| 2-16 years | ≤ 15 | gradually increase by approximately 10 mL/hr/site every 2-4 weeks up to a maximum of 25 |
* As tolerated
Administration/Handling instructions
CUTAQUIG is for subcutaneous use only.
Follow the administration guidance below and use aseptic technique when administrating CUTAQUIG.
 Figure 1 |
1. Getting ready for infusion — Choose and prepare a clean work area. — Gather your infusion equipment: Infusion pump and compatible syringe(s). Needle or needleless transfer device (for drawing up product from the vial). Infusion set (varies according to manufacturer’s instructions). Infusion tubing and Y-connector (if required). Ancillary supplies: disinfectant wipes, gauze or transparent dressing, tape and sharps container. Patient’s treatment diary/log book. — Wash your hands thoroughly and let them dry ( Figure 1 ). |
 Figure 2 |
2. Checking and opening the vials — If taken out of the refrigerator, allow products to reach room temperature (≤ 25°C / 77°F). — Inspect each vial carefully: Check that labelled dose is correct and based on prescription. Check that the expiration date has not been passed. Check the appearance of the solution: do not use the solution if it is cloudy or contains particles. Make sure the protective cap is not broken or missing. — Remove the protective cap. — Disinfect the rubber stopper by using a sterile wipe and allow it to dry ( Figure 2). |
 Figure 3 |
3. Preparing and filling the syringe — Open sterile syringe and needle or needleless transfer device. — If a needleless transfer device is used, follow the instructions of the device manufacturer. — If the transfer is done using needle and syringe follow the instructions below: Attach the needle to the syringe with a screw action. Draw back on the plunger to fill the syringe with air which should be roughly equal to the amount of solution needed from the vial. Insert the needle into the center of the vial stopper and turn the vial upside down. To avoid foaming, ensure that the tip of the needle is not in the solution, and then inject the air. Next, move the needle so that the tip is in the solution, and then slowly draw up the desired volume of CUTAQUIG ( Figure 3 ). Withdraw the needle from the vial. This procedure might be repeated if you need to use more than one vial. When finished, remove the needle and dispose of it into the sharps bin and proceed to the next step. |
 Figure 4 |
4. Preparing the infusion pump and tubing — Follow the manufacturer’s instructions for preparing the infusion pump. — Prime the administration tubing according to manufacturer’s instruction to eliminate all remaining air ( Figure 4 ). — Stop priming before fluid reaches the needle tip. |
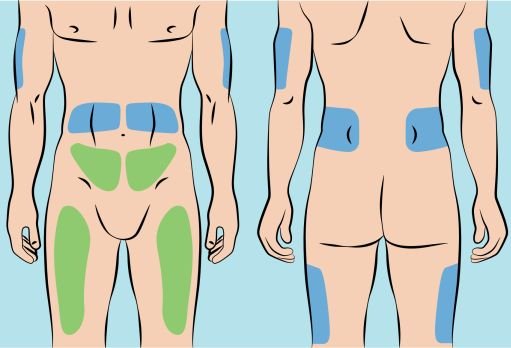 Figure 5 |
5. Preparing the infusion site(s) and inserting the infusion needle(s) — CUTAQUIG can be infused in the following areas: abdomen, thigh, upper arm, and/or upper leg/hip area ( Figure 5 ). — The number and location of injection sites depends on the volume of the total dose. The infusion sites should be at least 2 inches apart. A maximum of 6 infusion sites can be used simultaneously. — Rotate sites between infusions. — Avoid inserting the needle into scars, tattoos or injured/inflamed skin areas and inspect skin for signs of infection. — Clean your skin at your selected infusion site(s) with an antiseptic skin wipe starting at the center and working outward in a circular motion, and allow each site to dry before proceeding. |
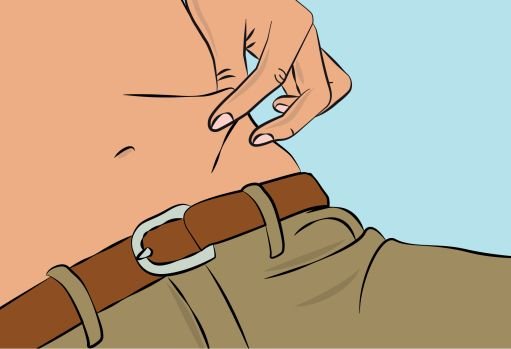 Figure 6 |
— Pinch the skin between your thumb and forefinger around the injection site ( Figure 6 ) and insert the needle into the subcutaneous tissue ( Figure 7 ). The angle of the needle will depend on the type of infusion set being used. |
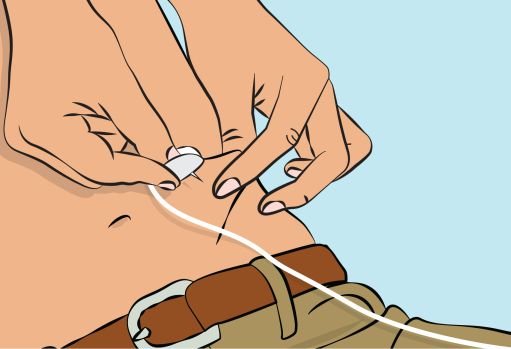 Figure 7 |
|
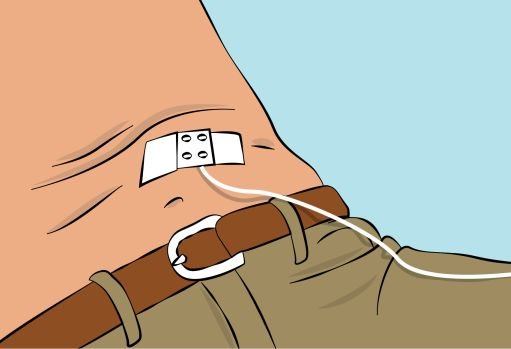 Figure 8 |
— Secure the needle in place by applying sterile gauze and tape or transparent dressing ( Figure 8 ). |
6. Checking the infusion
— Check needle placement by pulling back on the syringe plunger. There should not be any blood return in the tubing.
— If blood return is seen, remove needle and restart from step 5 with new tubing at a different location.
7. Starting the infusion
— Start the infusion. Follow infusion pump manufacturer’s instructions.
8. Recording the infusion
— On each vial of CUTAQUIG, you will find a peel-off portion of the label with the batch number details. Stick this label in your patient’s treatment diary or infusion log book. Record details of the dose, date, time, infusion site location and any infections, side effects or other comments.
9. After infusion is complete
— Gently remove the needle(s) and immediately place into the sharps bin.
— If necessary, press a small piece of gauze on the needle site and apply a dressing.
— Discard all used disposable supplies as well as any unused product and the empty vial(s) as recommended by your healthcare provider and according to local requirements.
— Clean and store the infusion pump according to the manufacturer’s instructions.
For self-administration, provide the patient and caregiver with instructions and appropriate training for infusion at home or other appropriate setting. Verify that the patient or caregiver is capable of self-administration or administration, respectively, using appropriate technique.
3. Dosage Forms and Strengths
CUTAQUIG is a solution containing 16.5% IgG (165 mg/mL).
4. Contraindications
CUTAQUIG is contraindicated:
- In patients who have had an anaphylactic or severe systemic reaction to the subcutaneous administration of human immune globulin or to any of the components of CUTAQUIG such as Polysorbate 80.
- In IgA-deficient patients with antibodies against IgA and a history of hypersensitivity to human globulin treatment.
5. Warnings and Precautions
5.1 Hypersensitivity
Severe hypersensitivity reactions may occur with CUTAQUIG, even in patients who tolerated previous treatment with human immune globulin. If a hypersensitivity reaction occurs, discontinue the CUTAQUIG infusion immediately and initiate appropriate treatment. Have epinephrine immediately available to treat any severe acute hypersensitivity reaction.
IgA-deficient patients with known anti-IgA antibodies have a higher risk of developing potentially severe hypersensitivity and/or anaphylactic reactions (including anaphylaxis and shock) with administration of CUTAQUIG. CUTAQUIG contains on average 0.206 mg of IgA /mL.
5.2 Thrombosis
Thrombosis may occur following treatment with immune globulin products, including CUTAQUIG. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in absence of known risk factors.
Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, such as those with cryoglobulins, fasting chylomicronemia/markedly high triglycerides, or monoclonal gammopathies. For patients at risk of thrombosis, administer CUTAQUIG at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity. [see
WARNING
,
Patient Counseling Information
(
17
)]
5.3 Falsely Elevated Glucose Readings
Blood Glucose Testing: Some types of blood glucose testing systems (for example, those based on the glucose dehydrogenase pyrroloquinolinequinone (GDH-PQQ) or glucose-dye-oxidoreductase methods) falsely interpret the maltose contained in CUTAQUIG as glucose. This may potentially result in falsely elevated glucose readings and, consequently, in the inappropriate administration of insulin, resulting in life-threatening hypoglycemia. Also, cases of true hypoglycemia may go untreated if the hypoglycemic state is masked by falsely elevated glucose readings. Accordingly, when administering CUTAQUIG, measure blood glucose with a glucose-specific method. The product information of the blood glucose testing system, including that of the test strips, should be carefully reviewed to determine if the system is appropriate for use with maltose-containing parenteral products. If any uncertainty exists, contact the manufacturer of the testing system to determine if the system is appropriate for use with maltose-containing parenteral products.
5.4 Aseptic Meningitis Syndrome (AMS)
Aseptic Meningitis Syndrome can occur with CUTAQUIG. AMS has been reported after the use of human immune globulin administered intravenously and subcutaneously. It usually begins a few hours to 2 days following treatment and occurs more frequently in females than in males.
AMS is characterized by the following signs and symptoms: severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea and vomiting. Cerebrospinal fluid (CSF) studies frequently show pleocytosis up to several thousand cells per mm
3
, predominantly from the granulocytic series, and elevated protein levels up to several hundred milligram/dL, but negative culture results. To rule out other causes of meningitis, conduct a thorough neurological examination on patients showing such symptoms and signs, including CSF studies. Discontinuation of immunoglobulin treatment has resulted in remission of AMS within several days without sequelae.
5.5 Renal Dysfunction/Failure
Acute renal dysfunction/failure, acute tubular necrosis, proximal tubular nephropathy, osmotic nephrosis and death may occur with use of human immune globulin, especially those containing sucrose. CUTAQUIG does not contain sucrose. Ensure that patients are not volume-depleted before administration of CUTAQUIG.
In patients at risk of developing renal dysfunction because of any degree of preexisting renal insufficiency or predisposition to acute renal failure (such as diabetes mellitus, age greater than 65 years, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs) monitor renal function and consider lower, more frequent dosing [see
Dosage and Administration
(
2
),
Patient Counseling Information
(
17
)].
Periodic monitoring of renal function and urine output is particularly important in patients judged to have a potential increased risk of developing acute renal failure. Assess renal function, including measurements of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of CUTAQUIG and again at appropriate intervals thereafter. If renal function deteriorates, consider discontinuing CUTAQUIG.
5.6 Hemolysis
IgG products, including CUTAQUIG, can contain blood group antibodies that may act as hemolysins and induce
in vivo
coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin (Coombs’) test result. Delayed hemolytic anemia can develop subsequent to immune globulin therapy due to enhanced RBC sequestration, and acute hemolysis, consistent with intravascular hemolysis, has been reported.
Monitor CUTAQUIG recipients for clinical signs and symptoms of hemolysis, particularly patients with risk factors (such as non-O blood group, administration of high IgG doses (≥ 2g/kg BW)). Underlying inflammatory state in a patient may increase the risk of hemolysis but its role is uncertain. Consider appropriate confirmatory laboratory testing if signs and symptoms of hemolysis are present after CUTAQUIG infusion.
5.7 Transfusion-related Acute Lung Injury (TRALI)
Non-cardiogenic pulmonary edema may occur in patients administered human immune globulin products. TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Typically, it occurs within 1 to 6 hours following transfusion.
Monitor patients for pulmonary adverse reactions. If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies in both the product and patient’s serum. Patients with TRALI may be managed using oxygen therapy with adequate ventilatory support.
5.8 Transmittable Infectious Agents
Because CUTAQUIG is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. This also applies to unknown or emerging viruses and other pathogens. No cases of transmission of viral diseases or CJD have been associated with the use of CUTAQUIG. All infections suspected by a physician to have possibly been transmitted by CUTAQUIG should be reported to Pfizer Inc. at 1-800-438-1985.
5.9 Interference with Laboratory Tests
After infusion of CUTAQUIG, the transitory rise of the various passively transferred antibodies in the patient’s blood may yield false positive serological test results, with the potential for misleading interpretation. Passive transmission of antibodies to erythrocyte antigens (e.g.. A, B, and D) may cause a positive direct or indirect antiglobulin (Coombs’) test.
6. Adverse Reactions/Side Effects
The most common adverse reactions (AR) observed in ≥ 5% of study subjects were local infusion site adverse reactions (such as redness, swelling, and itching), headache, fever, dermatitis, asthma, diarrhea, and cough.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical safety data are based on one pivotal open-label, single arm, prospective, multicenter major effectiveness study of CUTAQUIG in subjects with primary humoral immunodeficiency (PI), previously treated with Immune Globulin Intravenous (Human) (IGIV) for at least 6 months and its extension study. The pivotal study was conducted in Europe and North America and was followed by an extension safety study (prospective, open-label, single-arm, multicenter phase 3). 21 patients were initially treated in the pivotal study and 6 patients from Canada were newly treated. Total number of patients in both studies combined was 81.
In the pivotal study, the 75 subjects in the Safety Analysis Set received 4462 infusions, with a mean of 59 infusions administered per subject. The average dose of CUTAQUIG used per subject was 0.187 g/kg in adult subjects, 0.150 g/kg in young children, 0.164 g/kg in older children and 0.170 g/kg in adolescents.
In the extension study, the 27 subjects in the Safety Analysis Set received 2777 infusions. The average dose of CUTAQUIG used per subject was 0.166 g/kg in adult subjects, 0.127 g/kg in young children, 0.210 g/kg in older children and 0.160 g/kg in adolescents.
Excluding infections, a total of 74 out of 81 subjects (91%) experienced 1527 adverse reactions (ARs, defined as adverse events occurring during or within 72 hours of infusion or any adverse events otherwise causally related) in both studies. Of the 1527 ARs 1088 were infusion site reactions. The proportion of infusions with adverse reactions was 0.21. Sixty-one subjects (75%) had at least one systemic AR.
Local reactions were the most common ARs and were experienced by 59 subjects (73%). The rate of infusion site reaction per infusion was 0.15. Most local ARs were either mild (transient AR causing discomfort but not interfering with routine activities [87.5%]) or moderate (AR sufficiently discomforting to interfere with routine activities [10.3%]) in intensity.
Table 3
and
Table 4
summarize adverse reactions occurring in ≥ 5% of adult subjects and pediatric subjects in both the pivotal and the extension study.
Table 3 Adverse reactions* in ≥ 5% of adult subjects and rate per infusion in adult subjects (pivotal and extension study)
ARs |
Number (%) of subjects (N=43) |
Number (rate**) of ARs (N=3956 infusions) |
Local reaction |
31 (72.1) | 648 (16.4) |
Systemic ARs |
||
| Headache | 8 (18.6) | 10 (0.002) |
| Dermatitis | 5 (11.6) | 6 (0.001) |
| Fever | 5 (11.6) | 5 (0.001) |
| Diarrhoea | 5 (11.6) | 7 (0.002) |
| Muscle spasms | 4 (9.3) | 5 (0.001) |
| Back pain | 4 (9.3) | 4 (0.001) |
| Arthralgia | 3 (7.0) | 3 (0.001) |
* Excluding infections.
** Rate = total number of adverse reactions divided by total number of infusions.
Table 4 Adverse reactions* in ≥ 5% pediatric subjects and rate per infusion in pediatric subjects (pivotal and extension study)
ARs |
Number (%) of subjects (N=38) |
Number (rate**) of ARs (N=3283 infusions) |
Local reaction |
28 (73.7) | 442 (13.5) |
Systemic ARs |
||
| Asthma | 4 (10.5) | 7 (0.002) |
| Cough | 4 (10.5) | 5 (0.002) |
| Vomiting | 4 (10.5) | 5 (0.002) |
| Nasal congestion | 3 (7.9) | 4 (0.001) |
| Fever | 3 (7.9) | 4 (0.001) |
| Headache | 3 (7.9) | 3 (0.001) |
| Alanine aminotransferase increased | 3 (7.9) | 3 (0.001) |
| Leukopenia | 3 (7.9) | 3 (0.001) |
| Neutropenia | 3 (7.9) | 3 (0.001) |
| Dermatitis | 2 (5.3) | 3 (0.001) |
| Oropharyngeal pain | 2 (5.3) | 3 (0.001) |
| Urticaria | 2 (5.3) | 2 (0.001) |
| Aspartate aminotransferase increased | 2 (5.3) | 2 (0.001) |
| Abdominal pain | 2 (5.3) | 2 (0.001) |
| Ear pain | 2 (5.3) | 2 (0.001) |
* Excluding infections.
** Rate = total number of adverse reactions divided by total number of infusions.
The most frequent infusion site reactions were redness, swelling, and itching. During the pivotal study period the incidence of local reactions decreased over time from 37% of infusions triggering an infusion site reaction during the first 4 infusions to around 16% of infusions with an infusion site reaction during the last 4 infusions.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of immune globulin products administered subcutaneously. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Table 5
summarizes adverse reactions that have been reported during post-marketing use of immune globulin products:
Table 5 Adverse reactions reported during post-marketing use of immune globulin products
MedDRA System Organ Class (SOC) |
Adverse reactions |
| Blood and lymphatic system disorders | Pancytopenia, leukopenia, hemolytic anemia |
| Immune system disorders | Anaphylactic reaction, hypersensitivity reaction, allergic reaction, angioneurotic edema, face edema |
| Psychiatric disorders | Agitation |
| Nervous system disorders | Loss of consciousness, cerebrovascular accident, aseptic meningitis, seizures, migraine, tremor, paresthesia, dizziness |
| Cardiac disorders | Cardiac arrest, tachycardia, palpitations |
| Vascular disorders | Thromboembolism, thrombosis, circulatory collapse, hypertension, hematoma |
| Respiratory, thoracic and mediastinal disorders | Transfusion-related acute lung injury, acute respiratory distress syndrome, respiratory failure, pulmonary embolism, apnea, cyanosis, hypoxia, pulmonary edema, bronchospasm, dyspnea, cough, wheezing |
| Gastrointestinal disorders | Hepatic function abnormal, nausea, vomiting, abdominal distension, abdominal pain upper |
| Skin and subcutaneous tissue disorders | Stevens-Johnson syndrome, epidermolysis, erythema multiforme, eczema, urticaria, rash (erythematous), alopecia, skin discoloration, skin mass, skin reaction, skin/infusion site ulceration, skin/infusion site necrosis |
| Musculoskeletal and connective tissue disorders | Back pain, arthralgia, pain in extremity, myalgia |
| Renal and urinary disorders | (Acute) renal failure |
| General disorders and administration site condition | Chills, chest pain, chest discomfort, hot flush, flushing, hyperhidrosis, fatigue, influenza-like illness, malaise |
| Investigations | Hepatic enzyme increased, coombs test positive, free hemoglobin present, hemoglobin increased, haptoglobin decreased |
The following adverse reactions have been identified during post-approval use of CUTAQUIG: dizziness, nausea, pruritus and fatigue. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
To report SUSPECTED ADVERSE REACTIONS, contact Octapharma USA Inc. at 1-866-766-4860 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
7. Drug Interactions
7.1 Serological Testing
Various passively transferred antibodies in immunoglobulin preparations may lead to misinterpretation of results of serological testing.
7.2 Live Virus Vaccines
The passive transfer of antibodies with immunoglobulin administration may interfere with the response to live virus vaccines such as measles, mumps, rubella, and varicella. Inform the immunizing medical practitioner of recent therapy with CUTAQUIG so that appropriate precautions can be taken. [See
Patient Counselling Information
(
17
)]
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
No human data are available to indicate the presence or absence of drug-associated risk. Animal reproduction studies have not been conducted with CUTAQUIG. It is not known whether CUTAQUIG can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Immune globulins cross the placenta from maternal circulation increasingly after 30 weeks of gestation. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk summary
No human data are available to indicate the presence or absence of drug associated risk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for CUTAQUIG and any potential adverse effects on the breastfed infant from CUTAQUIG or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of CUTAQUIG have been established for primary humoral immunodeficiency in the pediatric age group 2 to 16 years CUTAQUIG was evaluated in 38 pediatric subjects with primary humoral immunodeficiency (26 children and 12 adolescents) in the pivotal and extension studies. [see
Clinical Studies
(
14
)]
Overall safety and efficacy findings in pediatric age group were comparable to those seen in adults. However, infusion site reactions were more commonly observed in adults compared to the pediatric population.
PK parameters are comparable between pediatric and adult patients. No pediatric-specific dose requirements were necessary to achieve the desired serum IgG levels.
Safety and effectiveness of CUTAQUIG in pediatric patients below the age of 2 have not been established.
8.5 Geriatric Use
Clinical studies of CUTAQUIG did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Three study subjects enrolled in the clinical trial were 65 years and over. In general, dose selection for an elderly patient should be cautious. Medical practitioners should start at the low end of the dosing range to reduce the risk of hyperviscosity and precipitation of cardiac, renal, or hepatic adverse reaction and of concomitant disease or other drug therapy.
11. Cutaquig Description
CUTAQUIG (Immune Globulin Subcutaneous (Human) — hipp), is a solvent/detergent (S/D)-treated, sterile preparation of highly purified immunoglobulin G (IgG) derived from large pools of human plasma. CUTAQUIG is a solution for injection to be administered subcutaneously.
This preparation contains approximately 165 mg of protein per mL (16.5%), of which not less than 96% is normal human immunoglobulin G. CUTAQUIG contains not more than 3% aggregates, not less than 94% monomers and dimers, and not more than 3% fragments. The product contains on average 0.206 mg of IgA /mL.
The sodium content of the final solution is not more than 30 mmol/L and the pH is between 5.0 and 5.5. The osmolality is 310 — 380 mOsmol/kg.
The manufacturing process for CUTAQUIG isolates IgG without additional chemical or enzymatic modification, and the Fc portion is maintained intact. CUTAQUIG contains the IgG antibody activities present in the donor population. IgG subclasses are fully represented with the following approximate percent of total IgG: IgG1 is 70%, IgG2 is 25%, IgG3 is 3% and IgG4 is 2%.
CUTAQUIG contains a broad spectrum of IgG antibodies against bacterial and viral agents that are capable of opsonization and neutralization of microbes and toxins. It contains maltose (79 mg/mL), but no preservatives or sucrose.
All units of human plasma used in the manufacture of CUTAQUIG are provided by FDA-approved blood and plasma establishments, and are tested by FDA-licensed serological tests for HBsAg, antibodies to HCV and HIV and Nucleic Acid Test (NAT) for HCV and HIV-1 and found to be non-reactive (negative).
The product is manufactured by the cold ethanol fractionation process followed by ultrafiltration and chromatography. The manufacturing process includes treatment with an organic S/D mixture composed of tri-n-butyl phosphate (TNBP) and Octoxynol. The CUTAQUIG manufacturing process shows significant viral reduction in in vitro studies (
Table 6
). These reductions are achieved through a combination of process steps including cold ethanol fractionation, S/D treatment and pH 4 treatment.
Table 6 Pathogen Reduction During CUTAQUIG Manufacturing
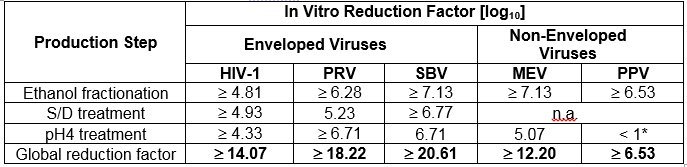
* Not calculated for global Log
10
Reduction Factor
HIV-1: Human Immunodeficiency Virus — 1
PRV: Pseudorabies Virus, model virus for e.g. Hepatitis B Virus (HBV)
SBV: Sindbis Virus, model virus for Hepatitis C Virus (HCV)
MEV: Mouse Encephalomyelitis Virus, model virus for Human Parvovirus B19
PPV: Porcine Parvovirus, model virus for Hepatitis A Virus (HAV)
n.a.: not applicable
12. Cutaquig — Clinical Pharmacology
12.1 Mechanism of Action
CUTAQUIG supplies a broad spectrum of opsonizing and neutralizing Immunoglobulin G (IgG) antibodies against a wide variety of bacterial and viral agents. It has a distribution of immune globulin subclasses closely proportional to that in native human plasma. The mechanism of action in primary humoral immunodeficiency (PI) has not been fully elucidated; however adequate doses may restore abnormally low immune globulin G levels to the normal range and thus help in preventing infections.
12.2 Pharmacodynamics
CUTAQUIG contains mainly IgG with a broad spectrum of antibodies against various infectious agents reflecting the IgG activity found in the donor population. CUTAQUIG which is prepared from pooled material from not less than 1000 donors, has an IgG subclass distribution similar to that of native human plasma. Adequate doses of Immune Globulin Subcutaneous (Human) (IGSC) can restore an abnormally low IgG level to the normal range. Standard pharmacodynamic studies were not performed.
12.3 Pharmacokinetics
A pharmacokinetic (PK) sub-study was conducted in 37 subjects (18 adults and 19 pediatric subjects) who were enrolled in the pivotal safety and efficacy study. Blood samples for PK study were collected prior to switching to CUTAQUIG (IGIV profile: PK
IV
), after the 11
th
infusion of CUTAQUIG (first SC profile: PK
SC1
) and after the 28th infusion of CUTAQUIG (second SC profile: PK
SC2
). The objective of the PK sub-study was to compare the AUCs following the IV and SC administration. At steady state, the ratio of geometric least square means (SC2:IV) was 1.06, (90% CI: 1.03, 1.10), indicating comparable exposure between IGSC and IGIV treatment (standardized to a 7-day period). The actual dose converting factor was 1.41 (1.21, 1.89). Using a Population PK model calculation, the DCF was determined in a statistically more advanced manner at 1.33 for a median patient.
Serum IgG and IgG subclass trough levels were nearly constant during the IGSC phase of the study, with higher mean levels after SC treatment compared with those following IGIV. At the end of the IGIV period, trough levels ranged from 5.0 g/L to 15.1 g/L. Over the entire IGSC treatment period individual trough levels of total IgG ranged between 4.4 g/L to 24.0 g/L.
Table 7
summarizes the Key PK parameters for CUTAQUIG. PK parameters are comparable between pediatric and adult patients.
Table 7 Key Pharmacokinetic Parameters for CUTAQUIG and IGIV
Parameter [Arithmetic Mean (SD)] |
IGIV (N=37) |
CUTAQUIG (N=37) |
|
C max [g/L] |
18.01 (4.5) | 13.47 (3.7) |
|
C min [g/L] |
10.09 (2.5) | 11.66 (2.9) |
|
T max [h] # |
3.38 (1.6 – 69.5) | 49.62 (0.8 – 98.3) |
|
AUC tau [g*hr/L] |
2013 (570)* | 2233 (586) |
|
AUC tau [mg*day/dL] |
8389 (2376)* | 9302 (2441) |
|
CL + (mL/day/kg) |
1.5 (0.5) | 1.9 (0.6) |
| Actual IgG Dose per kg Body Weight and Week (g/kg/week) | 0.121 (0.049) | 0.170 (0.066) |
#
T
max
is presented as Median (range)
* standardized to a 7-day period
+
apparent clearance (CL/F) for CUTAQUIG (F = bioavailability)
Compared with weekly administration, administration of CUTAQUIG on a every other week basis at double the weekly dose is expected to result in comparable IgG exposure [equivalent AUCs, with a slightly higher IgG peak (Cmax) and slightly lower trough (Cmin)] in both adult and pediatric subjects. In addition, for the same total weekly dose, CUTAQUIG infusions given 2, 3, 5, or 7 times per week (frequent dosing) are expected to produce IgG exposures comparable to weekly dosing in both adult and pediatric subjects.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies were conducted with CUTAQUIG to assess carcinogenesis, mutagenesis or impairment of fertility.
13.2 Animal Toxicology and/or Pharmacology
Safety of CUTAQUIG has been demonstrated in several standard nonclinical toxicology studies (local tolerance in rabbits, cardiovascular and respiratory effects in dogs, thrombogenic potential in rabbits).
The product given subcutaneously to CD1 mice inoculated with Streptococcus pneumoniae showed a dose related improvement in survival.
TNBP and Octoxynol may be found in CUTAQUIG in trace amounts. In animals and in vitro studies, these compounds were not genotoxic and did not have mutagenic properties. They did not show teratogenic effects when administered to pregnant rabbits and rats during organogenesis.
14. Clinical Studies
There were 2 clinical studies, one was a pivotal, prospective, open-label, single-arm, multicenter study to evaluate the pharmacokinetics (PK), efficacy, tolerability and safety of subcutaneous human immunoglobulin (CUTAQUIG) in subjects with primary humoral immunodeficiency (PI). The other was an extension study.
The pivotal study was conducted in 75 subjects (37 adult and 38 pediatric subjects < 17 years of age) who received weekly SC infusions with CUTAQUIG during a 12-week wash-in/wash-out period followed by a 12-month efficacy period during which efficacy, pharmacokinetics, safety, tolerability, and quality of life (QoL) parameters of CUTAQUIG were evaluated. All patients received study treatment and 68 patients completed the study. Seven patients (4 adolescents and 3 adults) were withdrawn prematurely. Reasons for withdrawal from the study were patient’s decision in 6 cases and patient’s noncompliance in one adolescent patient.
During the efficacy period the mean weekly dose was 174 mg/kg BW, with individual doses ranging from 60 to 390 mg/kg BW. The median duration of infusion per week was 1.5 hours.
All enrolled subjects (n=75) were included in the Safety Analysis Set and the Full Analysis Set (FAS). Four subjects were excluded from the Per-Protocol (PP) Set because they terminated early before the start of the primary treatment period.
Overall, 36 female subjects and 39 male subjects participated in this study. The youngest subject enrolled in the study was 2 years old and the oldest was 73 years old. The mean age in the adult group (17–75 yrs) was 47.5 years. The mean age at time of enrollment in the pediatric groups was 4.2 years, 7.9 years and 14.1 years in the 3 pediatric age groups [young children (ages 2-5 years), older children (ages 6-11 years) and adolescents (ages 12-16 years)] respectively. Reported race was white for all but one subject, and all subjects were of not Hispanic/Latino ethnicity.
The main objective of the pivotal study was to assess the efficacy of CUTAQUIG in preventing serious bacterial infections (SBI defined as bacteremia/sepsis, bacterial meningitis, osteomyelitis/septic arthritis, bacterial pneumonia and visceral abscess). This endpoint was considered successful if the upper bound of the one-sided 99% confidence interval for the rate of SBIs was < 1.0 per subject-year of follow up. This criterion was met, as no SBIs were reported at any time during the study.
Other endpoints of the pivotal study included, but were not limited to: the number of episodes of any other infections, along with type and severity of infection and time to resolution; number of days of use and annual rate of antibiotics; absence and number of days of absence from work/school/ kindergarten/day care; and hospitalizations due to infections and number of days and annual rate of hospitalisation.
Efficacy results of the pivotal study are summarized in
Table 8
.
Table 8 Summary of efficacy results of the pivotal study (FAS).
Adults |
Pediatrics |
Total |
|
Number of subjects (efficacy period) |
37 | 38 | 75 |
|
Total number of subject years Infections Annual rate [number of SBIs* per subject-year (upper one-sided 99% CI)] Annual rate [number of other infections per subject-year (two-sided 95% CI)] |
35.5
0 (0.13) |
35.0
0 (0.13) |
70.5
0 (0.06) |
|
Systemic antibiotic use Number of subjects (%) Annual rate [days on treatment per subject-year (two-sided 95%CI)] |
25 (67.6%) 32.1 (17.2, 60.1) |
24 (63.2%) 62.6 (31.0, 126.4) |
49 (65.3%) 47.2 (28.4, 78.6) |
|
Days out of work/school/kindergarten/day care due to infections Number of days Annual rate [days per subject-year (two-sided 95% CI)] |
72 2.0 (0.7, 5.7) |
180 5.2 (2.8, 9.4) |
252 3.6 (2.1, 6.0) |
|
Hospitalization due to infections Number of days Annual rate [days per subject-year (two-sided 95% CI)] |
0 0 0 (0.1) # |
4 29 0.8 (0.3, 2.6) |
4 29 0.4 (0.1, 1.3) |
* Defined as bacterial pneumonia, bacteremia/septicemia, osteomyelitis/septic arthritis, bacterial meningitis, and visceral abscess.
CI = confidence interval
#
calculation for the upper limit of two-sided 95% or one-sided 99% CI was based on standard Poisson model for zero counts
The extension study was a prospective, open-label, single-arm, multicenter phase 3 safety study that enrolled 27 patients (17 adults, 10 patients aged <17 years) with PI. Twenty-one patients were initially treated in the pivotal study and 6 patients were newly enrolled. Mean age was 39 years (range 6 to 73 years). Ten patients (37%) were male, all but 2 were White, none was Hispanic or Latino.
Patients received CUTAQUIG on a weekly (25 patients) or an “every other week” schedule (2 patients). The primary objective of this study was to assess the medium-to-long term safety and tolerability of CUTAQUIG. Secondary efficacy assessments included occurrence of SBIs, the annual rate of all infections of any kind or seriousness, hospitalizations due to infections, and antibiotic use. One adult had bacteremia/sepsis. The rate of SBI per person-year was 0.03 for adults, and 0.0 for all other age groups (overall rate of 0.018). Secondary efficacy results are summarized in
Table 9
.
Table 9 Summary of efficacy results of the extension study (FAS)
Adults |
Pediatrics |
Total |
|
Number of subjects |
17 | 10 | 27 |
|
Total number of subject years Infections Annual rate [number of SBIs* per subject year (upper limit of one-sided 99% CI)] Annual rate [number of other infections per subject-year (two-sided 95% CI)] |
33.4
0.03 (0.31) |
20.7
0 (0.22) |
54.1
0.02 (0.19) |
|
Systemic antibiotic use Number of subjects (%) Annual rate [treatment days per subject-year (two-sided 95% CI)] |
13 (76.5%) 30.9 (12.2, 78.4) |
6 (60.0%) 70.6 (22.7, 220.0) |
19 (70.4%) 46.0 (21.3, 99.4) |
|
Days out of work/school/kindergarten/day care due to infections Number of days Annual rate [days per subject-year (two-sided 95% CI)] |
55 1.7 (0.4 to 6.5) |
75 3.6 (1.3 to 10.3) |
130 2.4 (1.0, 5.5) |
|
Hospitalization due to infections Number of days Annual rate [days per subject-year (two-sided 95% CI)] |
3 10 0.3 (0.07 to 1.3) |
0 0 0 (0.2) # |
3 10 0.2 (0.04, 0.8) |
* Defined as bacterial pneumonia, bacteremia/septicemia, osteomyelitis/septic arthritis, bacterial meningitis, and visceral abscess.
CI = confidence interval
#
calculation for the upper limit of two-sided 95% or one-sided 99% CI was based on standard Poisson model for zero counts
16. How is Cutaquig supplied
CUTAQUIG is supplied in 1 g, 1.65 g, 2 g, 3.3 g, 4 g or 8 g single-use vials.
Carton NDC Number |
Container NDC Number |
Size |
Grams Protein |
|
0069-1061-02 0069-1802-02 0069-1476-02 0069-1960-02 0069-1509-02 0069-1965-02 |
0069-1061-01 0069-1802-01 0069-1476-01 0069-1960-01 0069-1509-01 0069-1965-01 |
6 mL 10 mL 12 mL 20 mL 24 mL 48 mL |
1 1.65 2 3.3 4 8 |
- Components used in the packaging of CUTAQUIG are not made with natural rubber latex.
- Store at +2°C to +8°C (36°F to 46°F) for up to 36 months from the date of manufacture. Within its shelf-life, the product may be stored at room temperature up to +25°C (77°F) for up to 9 months without being refrigerated again during this period, and must be discarded if not used after this.
- Keep the vial in the outer carton to protect it from light.
- Check the product expiration date on the vial label. Do not use beyond the expiration date.
- Do not freeze. Do not use frozen product.
- Do not mix with other products.
- Do not shake the vial.
- Use aseptic technique when preparing and administering CUTAQUIG. Prior to administration, visually inspect each vial for particulate matter, whenever the solution and container permit. Do not use if the solution is cloudy or contains particulates.
- The vial is for single-use only. Discard any unused product after each infusion, in accordance with local requirements.
17. Patient Counseling Information
Advise the patients to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Inform patients to immediately report the following signs and symptoms to their healthcare provider:
-
Hypersensitivity reactions to CUTAQUIG (including hives, generalized hives, tightness of the chest, wheezing, low blood pressure, and anaphylaxis): Advise patients to have epinephrine immediately available to treat any acute severe hypersensitivity reaction and ensure patients and their caregivers are properly trained in its use. [see
Warnings and Precautions
(
5.1
)]. -
Pain and/or swelling of an arm or leg with warmth over the affected area, discoloration of an arm or leg, unexplained shortness of breath, chest pain or discomfort that worsens on deep breathing, unexplained rapid pulse, or numbness or weakness on one side of the body [see
Warnings and Precautions
(
5.2
)]. -
Severe headache, neck stiffness, drowsiness, fever, sensitivity to light, painful eye movements, nausea, and vomiting [see
Warnings and Precautions
(
5.4
)]. -
Decreased urine output, sudden weight gain, fluid retention/edema, and/or shortness of breath [see
Warnings and Precautions
(
5.5
)]. -
Fatigue, increased heart rate, yellowing of the skin or eyes, and dark-colored urine [see
Warnings and Precautions
(
5.6
)]. -
Severe breathing problems, lightheadedness, drops in blood pressure, and fever [see
Warnings and Precautions
(
5.7
)].
Inform patients to be tested periodically to make sure they have the appropriate levels of IgG in their blood. These tests may result in adjustments to the CUTAQUIG dose.
Inform patients/caregivers that because CUTAQUIG is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent [see
Warnings and Precautions
(
5.8
),
Description
(
11
)].
Inform patients that CUTAQUIG may interfere with the response to live virus vaccines (e.g., measles, mumps, rubella, and varicella) and to notify their immunizing physician of recent therapy with CUTAQUIG [see
Drug Interactions
(
7.2
)].
Home treatment/Self administration
- If self-administration is deemed appropriate by the physician, it is highly important to clearly instruct the patient/caregiver and to provide a thorough training on subcutaneous administration of CUTAQUIG. The patient/caregiver needs to demonstrate the ability to independently administer subcutaneous infusions and document it.
- Ensure the patient/caregiver understands the importance of regular subcutaneous infusions to maintain appropriate steady state IgG levels.
- Instruct the patient/caregiver to keep a treatment diary/log where all information about each infusion should be clearly documented.
- Inform patients to interrupt or terminate the CUTAQUIG infusion if a hypersensitivity reaction occurs.
- Inform patients that they should be tested regularly to make sure they have the correct levels of CUTAQUIG (IgG) in their blood. These tests may result in adjustment to the CUTAQUIG dose.
- Instruct the patient/caregiver that local injection site reactions (such as swelling, redness, itching) are a common side effect of subcutaneous treatment, but also tell them to contact their healthcare provider if local reactions increase in severity or persist for more than a few days.
- Inform the patient that injection sites should be rotated between infusions and that CUTAQUIG is for subcutaneous infusion only.
Manufactured by:
Octapharma Pharmazeutika Produktionsges.m.b.H.
Oberlaaer Strasse 235
1100 Vienna, Austria
U.S. License No. 1646
Distributed by:
Pfizer Labs
Division of Pfizer Inc.
NY, NY 10017
PATIENT PACKAGE INSERT
Patient Information
CUTAQUIG
(kew’ ta kwig)
Immune Globulin Subcutaneous (Human) — hipp, 16.5% solution
Information for Patients
The following summarizes important information about CUTAQUIG. Please read it carefully before using CUTAQUIG and each time you get a refill, as there may be new information. This Patient Information does not take the place of talking with your healthcare provider about your medical condition or your treatment. If you have any questions after reading this, ask your healthcare provider.
What is CUTAQUIG?
CUTAQUIG is a ready-to-use liquid solution of Immunoglobulin G (IgG), also called antibodies, which protect the body against infection. CUTAQUIG is used to treat patients with primary humoral immunodeficiency (PI).
There are many forms of PI. The most common types of PI result in an inability to make a very important type of protein called antibodies, which help the body fight off infections from bacteria or viruses. Regular administration of CUTAQUIG will help your body to fight bacteria and viruses that cause infections. CUTAQUIG is made from human plasma that is donated by healthy people. CUTAQUIG contains antibodies collected from these healthy people; these antibodies replace the missing antibodies in PI patients.
Who should NOT use CUTAQUIG?
Do not use CUTAQUIG if you have ever had a severe allergic reaction to immune globulin or other blood products.
Tell your healthcare provider if you:
- ever had any severe reaction to other immune globulin medicines.
- were told that you have a condition called IgA deficiency.
- have a history of heart or blood vessel disease.
- have had blood clots or “thick blood”.
- have been immobile for some time.
What should I tell my healthcare provider before using CUTAQUIG?
Talk to your healthcare provider about any medical conditions that you have or have had.
Tell your healthcare provider that you are taking CUTAQUIG before you get vaccination as vaccines may not work while you are taking CUTAQUIG.
Tell your healthcare provider about all of the prescription and non-prescription medicines you take, including over-the-counter medicines, dietary supplements, or herbal medicines.
Tell your healthcare provider if you are pregnant or plan to get pregnant, or if you are nursing because CUTAQUIG might not be right for you.
Tell your healthcare provider if you have diabetes. If you need to do glucose testing, your healthcare provider may tell you to use a different way to monitor your blood sugar levels on the day that you receive a CUTAQUIG infusion. Some types of blood glucose testing systems (so called glucometers) falsely interpret the maltose contained in CUTAQUIG as glucose. If you are uncertain ask your healthcare provider which glucose testing system you can use while using CUTAQUIG.
How should I use CUTAQUIG?
CUTAQUIG is given under the skin (subcutaneously). This type of infusion can be given at home by yourself or by your caretaker after appropriate training. Administration of CUTAQUIG should be done at regular intervals. Only use CUTAQUIG by yourself after you have been instructed by your healthcare provider.
What are the possible or reasonably likely side effects of CUTAQUIG?
The most common side effects of CUTAQUIG are:
- Infusion site reactions (including but not limited to redness, swelling, itching, fluid in tissue, pain, mass, bruising)
- Headache
- Elevated body temperature
Call your healthcare provider or emergency department immediately if you have any of the following symptoms: difficulty breathing, chest tightness, itching, swelling of the face, rash, hives or dizziness. These could be signs of a serious allergic reaction.
Tell your healthcare provider immediately if you have any of the following symptoms. They could be signs of a serious drug reaction:
- Decreased urination, sudden weight gain, or swelling in the legs. These could be signs of a kidney problem.
- Bad headache with nausea, vomiting, stiff neck, fever, and sensitivity to light. These could be signs of meningitis, which is irritation of the lining around your brain.
- Pain, swelling, warmth, discoloration of an arm or leg, unexplained shortness of breath, chest pain, discomfort that worsens on deep breathing, unexplained rapid pulse, a lump in your legs or arms, or numbness or weakness on one side of the body. These could be signs of a blood clot.
- Chest pain or trouble breathing, or blue lips or extremities. These could be signs of a serious heart or blood problem.
- Brown or red urine, fast heart rate, yellow skin or eyes. These could be signs of a liver or heart problem.
- Fever over 100°F (38°C). This could be sign of an infection.
These are not all of the possible side effects from CUTAQUIG. Ask your healthcare provider for more information. You are encouraged to report side effects to Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088.
Talk to your healthcare provider about any side effect that bothers you or that does not go away. You can ask your healthcare provider for more information on possible side effects.
Whenever giving yourself treatments at home, make sure another responsible person is present to help treat side effects or help if a serious adverse reaction occurs. Ask your healthcare provider whether you should have rescue medications, such as antihistamines or epinephrine. If prescribed epinephrine for severe allergic reactions, make sure you receive adequate training from a healthcare provider for its proper use.
How should I store CUTAQUIG?
Keep CUTAQUIG in the outer carton to protect it from exposure to light.
Do not freeze CUTAQUIG.
Store CUTAQUIG at 36°F- 46°F (+2°C to +8°C) for up to 36 months from the date of manufacture. Within its shelf-life, the product can be stored at room temperature up to 77°F (up to +25°C) for up to 9 months without being refrigerated again during this period, and must be discarded if not used after this.
Do not use CUTAQUIG after the expiration date printed on the vial.
Dispose all materials, including any unused CUTAQUIG, in an appropriate container.
What else should I know about CUTAQUIG?
Do not use CUTAQUIG for a medical condition for which it was not prescribed. Do not share CUTAQUIG with other people, even if they have the same diagnosis and symptoms that you have.
Manufactured by
Octapharma Pharmazeutika Produktionsges.m.b.H.
Oberlaaer Strasse 235
1100 Vienna, Austria
U.S. License No. 1646
Distributed by
Pfizer Labs
Division of Pfizer Inc.
NY, NY 10017
CUTAQUIG
®
is a registered trademark of Octapharma.
Patient Instructions for Use
CUTAQUIG
Immune Globulin Subcutaneous (Human) — hipp, 16.5% solution
Detailed patient handling instructions for administration of CUTAQUIG
CUTAQUIG is for subcutaneous use only.
Use CUTAQUIG only after you have been properly instructed and trained by your healthcare provider.
Follow the administration guidance below step by step and use aseptic/sterile technique when administrating CUTAQUIG. Use gloves if you have been told to do so when preparing the infusion.
1. Prepare the necessary number of CUTAQUIG vials — If stored in the refrigerator, put the vials at room temperature at least 90 minutes prior to infusion. — Do not heat the vials or put them into the microwave. — Do not shake the vials to avoid foaming. |
|
 Figure 1 |
2. Getting ready for infusion — Choose and prepare a clean work area using antiseptic wipes or disinfecting solution. — Gather your infusion equipment: — Infusion pump and compatible syringe(s) Needle or needleless transfer device (for drawing up product from the vial) Infusion set (varies according to manufacturer’s instructions) Infusion tubing and Y-connector (if required) Ancillary supplies: disinfectant wipes, gauze or transparent dressing, tape and sharps container Treatment diary and pen — Wash and clean your hands thoroughly and let them dry as has been shown to you during the training ( Figure 1 ). You can wear gloves during the preparation of the infusion if you have been told so during the training. — If necessary, program the pump according to the user manual and as you have been shown during the training by your healthcare provider. |
 Figure 2 |
3. Checking and opening the vials — Allow products to reach room temperature (77°F / ≤ 25°C). — Inspect each vial carefully: Check that the labelled dose is correct and based on your prescription. Check that the expiration date has not been passed. Check the appearance of the solution (it should be clear and colorless to pale yellow or slightly opalescent). Do not use the solution if it is cloudy or contains particles. Make sure the protective cap is not broken or missing. — Remove the protective cap. — Disinfect the rubber stopper by using a sterile wipe and allow it to dry ( Figure 2 ). |
 Figure 3 |
4. Preparing and filling the syringe — Open sterile syringe and needle or needleless transfer device. — If a needleless transfer device is used, follow the instructions of the device manufacturer. — If the transfer is done using needle and syringe, follow the instructions below: Attach the needle to the syringe with a screw action. Draw back on the plunger to fill the syringe with air. The amount of air should be roughly equal to the amount of solution needed from the vial. Insert the needle into the center of the vial stopper and slowly turn the vial upside down. To avoid foaming, ensure that the tip of the needle is not in the solution; then inject air by pushing the plunger of the syringe. Next, move the needle so that the tip is in the solution; then slowly draw up the desired volume of CUTAQUIG solution, making sure that the needle tip is always in the solution ( Figure 3 ). Withdraw the needle from the vial. This procedure might be repeated if you need multiple vials for the calculated dose. When finished, remove the needle and dispose of it into the sharps bin. Once finished, proceed to the next step. |
 Figure 4 |
5. Preparing the infusion pump and tubing — Follow the manufacturer’s instructions for preparing the infusion pump. — To prime the administration tubing, attach the filled syringe to the infusion tubing and gently push the plunger to fill the tubing with CUTAQUIG, as has been shown to you during training ( Figure 4 ). — Stop priming before fluid reaches the needle tip. |
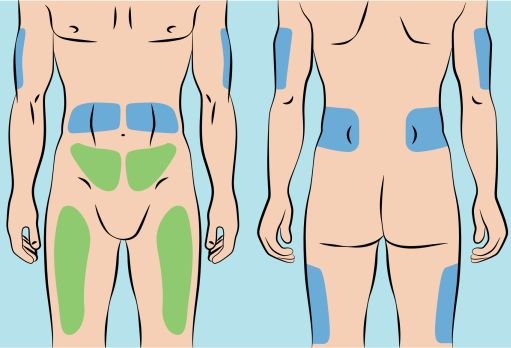 Figure 5 |
6. Preparing infusion site(s) and inserting the infusion needle(s) — CUTAQUIG can be infused into the following areas: abdomen, thigh, upper arm, and/or upper leg/hip area ( Figure 5 ). — The number and location of injection sites depends on the volume of the total dose. — The infusion sites should be at least 2 inches apart. Do not use more than 6 infusion sites at the same time. — Rotate sites between infusions. — Avoid inserting the needle into scars, tattoos, stretch marks or any skin that has signs of infection (such as injured/inflamed/red skin area). — Clean your skin at your selected infusion site(s) with an antiseptic skin wipe, starting at the center and working outward in a circular motion. Allow each site to dry before proceeding. |
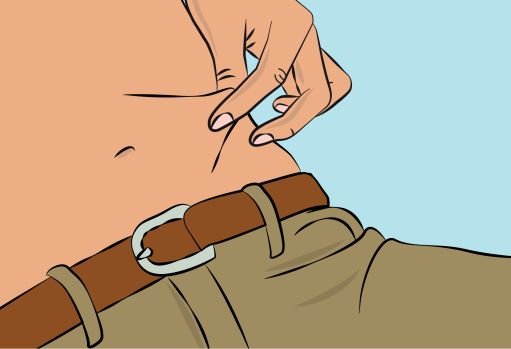 Figure 6 |
— Pinch the skin between your thumb and forefinger around the injection site ( Figure 6 ). Carefully remove the needle cover and insert the needle into the skin ( Figure 7 ). The angle of the needle will depend on the type of infusion set being used. — Put transparent dressing or sterile tape and gauze over the administration site to keep the needle in place during the infusion. |
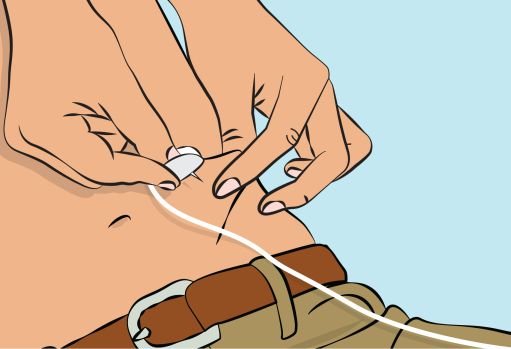 Figure 7 |
|
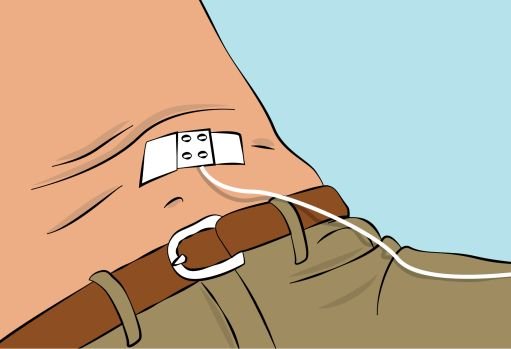 Figure 8 |
7. Checking the infusion — Check needle placement by pulling back on the syringe plunger. There should not be any blood return in the tubing ( Figure 8 ). — If blood return is seen, remove the needle and restart from step 6 with new tubing at a different location. |
8. Starting the infusion
— Start the infusion. Follow infusion pump manufacturer’s instructions.
9. Recording the infusion
— On each vial of CUTAQUIG, you will find a peel-off portion of the label with the batch number details. Stick this label in your patient’s treatment diary or infusion log book. Record details of the dose, date, time, infusion site location and any infections, side effects or other comments in connection with this infusion.
10. After infusion is complete
— Gently remove the dressing and the needle(s) and immediately place into the sharps bin.
— Press a small piece of gauze on the needle site and apply a dressing.
— Discard all used disposable supplies as well as any unused product and the empty vial(s) as recommended by your healthcare provider and according to local requirements.
— Tidy up and securely store all the reusable equipment (e.g., pump) until the next infusion.
If you encounter any problems or experience side effects during or after the infusion, contact your healthcare provider. When doing so, keep your treatment diary or log book with you to be able to give all necessary information.
You can also report side effects to the FDA at 1-800-FDA-1088 or online under www.fda.gov/medwatch.
Manufactured by
Octapharma Pharmazeutika Produktionsges.m.b.H.
Oberlaaer Strasse 235
1100 Vienna, Austria
U.S. License No. 1646
Distributed by
Pfizer Labs
Division of Pfizer Inc.
NY, NY 10017
CUTAQUIG
®
is a registered trademark of Octapharma.
Issued 11 2021.
PACKAGE LABEL — PRINCIPAL DISPLAY PANEL
Immune Globulin Subcutaneous (Human) — hipp, 16.5%
Cutaquig
6 mL
NDC 0069-1061-02

Cutaquig
10 mL
NDC 0069-1802-02
Cutaquig
12 mL
NDC 0069-1476-02
Cutaquig
20 mL
NDC 0069-1960-02
Cutaquig
24 mL
NDC 0069-1509-02
Cutaquig
48 mL
NDC 0069-1965-02
КУТАКВИГ
| Международное непатентованное наименование | Immunoglobulins, normal human, for extravascular adm. |
| АТС-код | J06BA01 |
| Тип МНН | Моно |
| Форма выпуска |
раствор для инъекций, 165 мг/мл; по 6 мл, 10 мл, 12 мл, 20 мл, 24 мл, 48 мл во флаконе, по 1 флакону в картонной коробке |
| Условия отпуска |
по рецепту |
| Состав |
1 мл раствора для инъекций содержит 165 мг иммуноглобулина человека нормального, /соответствующего содержанию общего белка, содержащего > 95 % IgG распределение подклассов IgG (приблизительные значения): IgG1 – 71 %, IgG2 – 25%, IgG3 – 3%, /IgG4 – 2%. Максимальное содержание IgA – не более 600 мкг/мл |
| Фармакологическая группа | Иммунные сыворотки и иммуноглобулины. Иммуноглобулин человека нормален для экстраваскулярного введения. |
| Заявитель |
Октафарма Фармацевтика Продуктіонсгес. м.б.Х. Австрия |
| Производитель 1 |
ОКТАФАРМА АБ(виробник, відповідальний за виробництво in-bulk, первинну упаковку, контроль якості, випуск серії) Швеция |
| Производитель 2 |
Октафарма Фармацевтика Продуктіонсгес. м.б.Х (виробник, відповідальний за виробництво in-bulk, первинну упаковку, контроль якості, візуальну інспекцію, маркування, вторинну упаковку, випуск серії) Австрия |
| Производитель 3 |
Октафарма Дессау ГмбХ (виробник, відповідальний за візуальну інспекцію, маркування, вторинну упаковку) Німеччина |
| Регистрационный номер | UA/20059/01/01 |
| Дата начала действия | 08.06.2023 |
| Дата окончания срока действия | 08.06.2028 |
| Досрочное прекращение | Нет |
| Срок годности | 3 года. После первого открытия флакона лекарственное средство следует использовать немедленно. |
Состав
Действующее вещество иммуноглобулин человека нормальный;
1 мл раствора для инъекций содержит 165 мг иммуноглобулина человека нормального,
Что соответствует содержанию общего белка, содержащего > 95% IgG;
Распределение подклассов IgG (приблизительные значения) IgG1
– 71%, IgG2
– 25%, IgG3
– 3%,
IgG4
– 2% Максимальное содержание IgA
– не более 600 мкг/мл;
Другие составляющие мальтоза, полисорбат 80, вода для инъекций
Лекарственная форма
Раствор для инъекций
Основные физико-химические свойства прозрачная и бесцветная жидкость, которая при хранении может стать слегка опалесцирующей или бледно-желтой
Фармакотерапевтическая группа
Иммунные сыворотки и иммуноглобулины Иммуноглобулин человека нормален для экстраваскулярного введения Код ATX J06B A01
Иммунологические и биологические свойства
Фармакодинамика
Иммуноглобулин человека нормальный содержит в основном иммуноглобулин G (IgG) с широким спектром антител к возбудителям инфекционных заболеваний
Иммуноглобулин человека нормальный содержит антитела к IgG, которые имеются в здоровой популяции Его обычно получают из пулов плазмы не менее 1000 доноров Он имеет распределение подклассов IgG, почти пропорциональное распределению в природной плазме крови человека Соответствующие дозы этого лекарственного препарата могут восстанавливать очень низкие уровни иммуноглобулина G до нормального диапазона значений
В клиническом исследовании в общей сложности 75 пациентов с синдромами первичного иммунодефицита проходили лечение препаратом Кутаквиг в течение периода до 64 недель В среднем еженедельная доза на одного пациента составляла 0,187 г/кг у взрослых пациентов, 0,150 г/кг у младшего возраста, 0,164 г/кг у детей старшего возраста и 0,170 г/кг у подростков Пациенты получали в общей сложности 4462 инъекции препарата Кутаквиг еженедельно
О серьезных бактериальных инфекциях не сообщалось ни во время периода накопления/выведения из организма, ни во время периода влияния препарата у пациентов, получавших Кутаквиг в течение клинического исследования
Пациенты детского возраста
Никакой разницы в фармакодинамических свойствах препарата не наблюдалось между взрослыми и пациентами детского возраста
Фармакокинетика
В клиническом исследовании III фазы фармакокинетическое (ФК) подследование проводилось с участием 37 пациентов с синдромом первичного иммунодефицита (СПИ) Образцы крови для ФК исследования отбирали перед переходом на препарат Кутаквиг (профиль ФКIV), после 11-й инъекции препарата Кутаквиг (первый ПШ профиль ФКпш1) и после 28-й инъекции препарата Кутаквиг (второй ПШ профиль ФКпш2)
Цель ФК подследствия заключалась в том, чтобы сравнить площади под ФК кривой (AUCs) после внутривенного (ВВ) и подкожного (ПЖ) введения, используя коэффициент коррекции дозы (КПД) 1,5 С помощью ФК модели популяции оценивались параметры ФК и проводились имитации
Абсорбция и распределение
После подкожного введения препарата Кутаквиг максимальные уровни в сыворотке крови достигаются примерно через 2 дня
Благодаря постепенной абсорбции подкожное введение иммуноглобулина (ПШВИГ) приводит к более плоским профилям и более слабым колебаниям в равновесном состоянии по сравнению с таковыми при внутривенном введении иммуноглобулина (ВВВИГ) средняя максимальная концентрация Сmax была ниже после ПШВИГ(13,2 + 3 л и 13,5 + 3,7 г/л для ФКпш1 и ФKпш2 соответственно), чем после ВВВИГ (18,0 + 4,5 г/л)
Соответственно, средние минимальные уровни сывороточного IgG и подкласса IgG были выше после ПЖ введения (11,5 и 11,7 г/л для ФКпш1 и ФKпш2 соответственно; общий диапазон составлял от 6,5 до 18,9 г/л) по сравнению с общим диапазоном в конце периода ВВВИГ (10,1 г/л; диапазон от 6,5 г/л до 14,3 г/л) Подсчитанная биодоступность при ПЖ введении составляла 75%, что соответствовало коэффициенту коррекции дозы 1,3 на основе массы тела для достижения одинаковой экспозиции AUC после ПШВИГ и ВВВИГ
ФК моделирования и имитации, проводимых на основе данных, полученных в клиническом исследовании с еженедельным введением дозы препарата Кутаквиг, указывают на то, что доза, скорректированная в соответствии с массой тела без КД для низшей биодоступности при ПЖ введении, является достаточной для поддержания системного воздействия IgG в терапевтическом диапазоне при интервалах введения до 1 недели, а также для введений более частых, чем один раз в неделю (например ежедневных) Более длинные интервалы дозировок (особенно при более низких исходных уровнях IgG) увеличивают риск падения IgG ниже минимального уровня 5 г/л
Пример Допуская исходный уровень IgG 4,0 г/л и переход с ВВВИГ к ПШВИГ с коэффициентом отношения доз 1,0 предполагалось, что доля пациентов с падением IgG ниже минимального уровня 5 г/л увеличится на 4 % при интервале дозировок 2 недели по сравнению с 1,4 % при интервалах дозировок ≤ Q1W (один раз в неделю)
Выведение из организма
IgG и IgG-комплексы разрушаются в клетках ретикулоэндотелиальной системы Было подсчитано, что средний период полувыведения IgG после введения препарата Кутаквиг у пациентов с СПИ составляет примерно 16 (9,2– 36,3) дней, как подсчитано в ФК модели популяции, при этом предполагалось нулевое (отсутствующее) эндогенное продуцирование IgG
Пациенты детского возраста
Никакой клинически значимой разницы не наблюдалось между фармакокинетическими параметрами у взрослых испытуемых и исследуемых пациентов детского возраста с СПИ
ФК моделирования и имитация, проводимых на основе данных, полученных в клиническом исследовании с еженедельным введением дозы препарата Кутаквиг, указывают на то, что доза, скорректированная в соответствии с массой тела, является достаточной для поддержания системного воздействия IgG в терапевтическом диапазоне, что не зависит от возраста
Доклинические данные по безопасности
Иммуноглобулины являются обычными составляющими плазмы крови человека Доклинические данные не обнаружили особой опасности для людей на основании обычных доклинических исследований фармакологической безопасности и местной переносимости
Поскольку клинический опыт не дает подтверждения канцерогенного или мутагенного потенциала иммуноглобулинов, никакие экспериментальные исследования с участием гетерогенных видов не проводились
Показания
Как заместительная терапия для взрослых и детей (возрастом 0– 18 лет) при таких состояниях
Синдром первичного иммунодефицита (СПИ) с пониженным образованием антител (см раздел «Особенности применения»); вторичные иммунодефициты при тяжелых или повторных инфекциях, когда лечение антимикробными препаратами неэффективно и установлена недостаточность специфических антител (ВНСА)* или уровни сывороточного IgG 4г/л
* ВНСА– неспособность достичь по крайней мере 2-кратного повышения титра антител IgG к вакцинам пневмококкового полисахарида и полипептидного антигена
Противопоказания
Повышенная чувствительность к действующему веществу или любому из вспомогательных веществ препарата, перечисленных в разделах «Состав» и «Особенности применения»
Кутаквиг нельзя вводить внутрисосудисто
Препарат также нельзя вводить внутримышечно при тяжелой тромбоцитопении и других нарушениях гемостаза
Взаимодействие с другими лекарственными средствами и другими видами взаимодействий
Живые противовирусные вакцины
Введение иммуноглобулина может ослабить на период от 6 недель до 3 месяцев эффективность живых противовирусных вакцин против таких инфекционных заболеваний, как корь, краснуха, эпидемический паротит (свинка) и ветряная оспа Интервал между введением этого лекарственного средства и вакцинацией живыми противовирусными вакцинами должен составлять 3 месяца В случае вакцинации против кори такое ослабление может занять до 1 года Поэтому у пациентов, получающих вакцину против кори, необходимо проверить состояние антител
Исследование содержания сахара в крови
Кутаквиг содержит мальтозу, которая может быть неверно истолкована (воспринята) как глюкоза определенными видами систем определения содержания глюкозы в крови Из-за возможности ошибочно увеличенных показателей глюкозы следует использовать для исследования или контроля уровня глюкозы в крови пациентов с сахарным диабетом только те системы, которые предназначены специально для измерения уровня глюкозы в крови
Пациенты детского возраста
Перечисленные взаимодействия касаются как взрослых, так и детей
Особенности применения
Лекарственное средство изготовлено из плазмы крови человеческих доноров
В каждом флаконе по 6 мл содержится 1 г иммуноглобулина нормального человека
В каждом флаконе по 10 мл содержится 1,65 г иммуноглобулина нормального человека
В каждом флаконе по 12 мл содержится 2 г иммуноглобулина человека нормального
В каждом флаконе по 20 мл содержится 3,3 г иммуноглобулина нормального человека
В каждом флаконе по 24 мл содержится 4 г иммуноглобулина нормального человека
В каждом флаконе по 48 мл содержится 8 г иммуноглобулина нормального человека
Настоятельно рекомендуется при введении препарата Кутаквиг каждый раз записывать название и номер серии препарата для того, чтобы можно было проследить связь между состоянием пациента и введением препарата конкретной серии
Кутаквиг применяется только для подкожного введения Если случайно ввести этот препарат в кровеносный сосуд, у пациента может развиться шок
Необходимо строго соблюдать рекомендованную скорость инфузии, указанную в разделе «Способ применения и дозы» По состоянию пациента необходим строгий контроль возникновения любых симптомов во время всего периода введения препарата
Некоторые побочные реакции могут возникать чаще у пациентов, получающих нормальный иммуноглобулин человека впервые или в редких случаях при переходе из заместительных препаратов на иммуноглобулин человека нормальный, а также в случае длительного перерыва после предварительной инъекции
Возможных осложнений часто можно избежать, если
Сначала медленно вводить препарат (см раздел «Способ применения и дозы»); убедиться в том, что за пациентами осуществляется строгий контроль выявления любых симптомов в течение всего времени введения инъекции В частности, пациенты, ранее не лечившиеся иммуноглобулином человека нормальным, пациенты, перешедшие из альтернативного иммуноглобулинового препарата, и пациенты, у которых прошел длительный промежуток времени с момента предварительной инъекции, должны находиться под пристальным наблюдением во время первой инъекции и в течение часа после первой инъекции для того, чтобы своевременно выявить нежелательные симптомы Все остальные пациенты должны находиться под наблюдением не менее 20 минут после введения препарата
В случае развития побочной реакции необходимо снизить скорость введения или прекратить инъекцию Если подозрение на аллергическую или анафилактическую реакцию, следует немедленно прекратить инъекцию Требуемое лечение зависит от характера и тяжести побочной реакции При шоке следует провести стандартную медицинскую противошоковую терапию
Гиперчувствительность
Истинные аллергические реакции возникают редко Они наиболее возможны у пациентов с антителами к иммуноглобулину А (IgA), которых следует лечить с чрезвычайной осторожностью Пациенты с антителами к IgA, для которых подкожное применение препаратов IgG остается единственным вариантом лечения, должны проходить терапию препаратом Кутаквиг только под тщательным контролем
Иногда иммуноглобулин человека нормальный может вызвать падение АД с анафилактической реакцией даже у пациентов, прошедших предварительное лечение иммуноглобулином человека нормальным
Тромбоэмболия
Артериальные и венозные осложнения тромбоэмболические, такие как инфаркт миокарда, инсульт, глубокий тромбоз вен и эмболия легких, связанные с использованием иммуноглобулинов Пациенты должны иметь достаточное содержание жидкости в организме перед применением иммуноглобулинов Следует соблюдать осторожность в отношении пациентов с предварительно существующими факторами риска тромботических осложнений (такими как пожилой возраст, артериальная гипертензия, сахарный диабет и наличие в анамнезе сосудистых заболеваний или тромботических осложнений, приобретенные или унаследованные тромбофилические нарушения, длительные периоды недвижимости, тяжелая гиповол вязкость крови)
Пациентам следует информировать о первых симптомах тромбоэмболических осложнений, таких как затрудненное дыхание, боль и отек конечности, очаговые неврологические симптомы и боль в груди, и посоветовать им немедленно обратиться к врачу при возникновении таких симптомов
Синдром асептического менингита (САМ)
Сообщалось о синдроме асептического менингита, возникавшем в связи с лечением иммуноглобулином, который вводится подкожно; симптомы обычно начинаются в течение периода от нескольких часов до 2 дней после лечения Прекращение лечения иммуноглобулином может привести к исчезновению САМ в течение нескольких дней без каких-либо последствий
Пациентам следует информировать о первых симптомах, включающих сильную головную боль, скованность затылка (шеи), сонливость (вялость), лихорадку, фотофобию, тошноту и рвоту
Дисфункция почек/ почечная недостаточность
Сообщалось о случаях тяжелых побочных реакций со стороны почек у пациентов, получавших лечение иммуноглобулином, особенно препаратами, содержащими сахарозу (препарат Кутаквиг не содержит сахарозы) К тяжелым побочным реакциям относятся ОПН, острый канальцевый некроз, проксимальная канальцевая нефропатия и осмотический нефроз
Факторы, увеличивающие риск почечных осложнений, включают, в частности, предварительно существующую почечную недостаточность, сахарный диабет, гиповолемию, сопутствующую терапию нефротоксическими лекарственными препаратами, возраст старше 65 лет, сепсис, повышенную вязкость и парапротеинемию
Гемолиз
Препараты IgG могут содержать антитела групп крови, которые могут действовать как гемолизины и вызывать in vivo покрытие эритроцитов иммуноглобулином, давая положительный результат прямого антиглобулинового теста (прямой пробы Кумбса) и редко могут вызывать гемолиз
Необходимо контролировать состояние реципиентов иммуноглобулиновых препаратов относительно клинических признаков и симптомов гемолиза
Влияние на серологическое исследование
После введения иммуноглобулина транзиторное увеличение количества различных пассивно поступающих в кровь пациента антител может привести в результате к получению ошибочно положительных результатов при серологическом исследовании
Пассивное поступление антител в эритроцитарные антигены, например А, B, D, может влиять на некоторые серологические исследования антиэритроцитарных антител, такие как прямой антиглобулиновый тест (DAT, прямая проба Кумбса)
Передача микроорганизмов
Стандартные меры по профилактике и предупреждению инфекций, возникающих в результате использования лекарственных препаратов, изготовленных из крови или плазмы человека, включают отбор доноров, скрининг индивидуальных порций донорской крови и пулов плазмы для выявления специфических маркеров инфекций, а также использование в процессе производства эффективных методов инактивации /удаление вирусов Несмотря на это, когда назначают лекарственные препараты из крови или плазмы человека, нельзя полностью исключить возможность передачи возбудителей инфекций Это также относится к неизвестным или новым вирусам и другим патогенным микроорганизмам
Принимаемые меры считаются эффективными в отношении оболочковых вирусов, таких как вирус иммунодефицита человека (ВИЧ), вирус гепатита В (ВГВ), вирус гепатита С (ВГС)
Эффективность принятых мер может быть ограничена в отношении безоболочковых вирусов, таких как вирус гепатита А (ВГА) и парвовирус В19
Существует обнадеживающий клинический опыт невозможности передачи гепатита А или парвовируса В19 вместе с иммуноглобулинами, также предположено, что содержание антител является важной составляющей вирусной безопасности
Пациенты детского возраста
Перечисленные особые указания и меры предосторожности касаются как взрослых, так и детей
Содержимое мальтозы
Этот лекарственный препарат содержит максимум 90 мг мальтозы в качестве вспомогательного вещества на 1 мл раствора Влияние мальтозы при анализах на определение содержания глюкозы в крови может привести к ошибочно увеличенным показателям глюкозы и, соответственно, к нецелесообразному (недопустимому) введению инсулина, что может стать причиной угрожающей жизни гипогликемии и иметь летальное последствие Также случаи настоящей гипогликемии могут остаться нелеченными, если гипогликемическое состояние маскируется по ошибочно увеличенным показателям глюкозы (см раздел «Взаимодействие с другими лекарственными средствами и другие виды взаимодействий»)
Содержание натрия
Этот лекарственный препарат содержит 33,1 мг натрия на флакон из 48 мл раствора для инъекций и 13,8 мг натрия на флакон с 20 мл раствора для инъекций, что эквивалентно 1,7% и 0,7% соответственно рекомендованной ВОЗ максимальной суточной нормы потребления натрия для взрослого, составляющей 2 г
Особенности применения и утилизации
Препарат необходимо довести до комнатной температуры или температуры тела перед использованием
Препарат следует осмотреть (проверить) визуально на наличие механических примесей (твердых частиц) и обесцвечивание перед применением
Растворы, помутневшие или содержащие осадок, нельзя использовать
Любой неиспользованный препарат или отходы следует утилизировать в соответствии с требованиями местных органов
Применение в период беременности и кормлении грудью
Беременность
Безопасность применения этого лекарственного средства беременным женщинам не была установлена в контролируемых клинических исследованиях, поэтому его следует назначать с осторожностью беременным и кормящим грудью Как оказалось, иммуноглобулиновые препараты проникают в плаценту в большей степени в течение третьего триместра беременности Клинический опыт применения иммуноглобулинов указывает на то, что не следует ожидать вредного влияния на течение беременности, плода или новорожденного
Кормление грудью
Иммуноглобулины выделяются в грудное молоко и могут способствовать защите младенца от патогенных микроорганизмов, проникающих через слизистую
Фертильность
Клинический опыт применения иммуноглобулинов свидетельствует о том, что не следует ожидать вредного влияния на фертильность
Способность влиять на скорость реакции при управлении транспортом или другими механизмами
Влияние на способность управлять транспортными средствами и работать с другими механизмами может снижаться при возникновении нежелательных реакций, связанных с препаратом Кутаквиг Пациенты, у которых отмечаются побочные реакции во время лечения, должны подождать до полного и окончательного исчезновения перед тем, как управлять транспортными средствами и работать с другими механизмами
Способ применения и дозы
Заместную терапию следует начать и контролировать под наблюдением врача, имеющего опыт лечения иммунодефицита
Дозы
Доза и режим дозировки зависит от показаний
Заместительная терапия
Этот препарат следует вводить подкожно
При заместительной терапии дозу подбирают индивидуально для каждого пациента в зависимости от фармакокинетической характеристики и клинической реакции (ответы на лечение) Кутаквиг можно вводить с постоянными интервалами, начиная с ежедневного введения и заканчивая введением один раз в две недели Представленные ниже режимы дозы препарата предоставляются в качестве рекомендаций
Режим дозирования должен обеспечить достижение минимального уровня IgG (определяющегося перед последующей инфузией) не менее 5–6 г/л и способствовать нахождению этого показателя в пределах референтных значений сывороточного IgG в соответствии с возрастом пациента Может оказаться необходимой ударная (насыщающая) доза не менее 0,2–0,5 г/кг (1,2–3,0 мл/кг) массы тела Может потребоваться разделить ее на несколько введений, которые осуществлять в течение нескольких дней, при этом максимальная суточная доза составляет 0,1–0,15 г/кг
Заместительная терапия при синдромах первичного иммунодефицита (как приведено в разделе «Показания»)
После достижения устойчивых уровней IgG поддерживающие дозы вводятся с одинаковыми повторными интервалами для достижения кумулятивной (суммарной) ежемесячной дозы примерно 0,4–0,8 г/кг (2,4–4,8 мл/кг) Для каждой отдельной дозы может потребоваться другой анатомический участок (место)
Минимальные уровни IgG следует измерять и оценивать вместе с частотой инфекции Чтобы уменьшить частоту инфекции, может потребоваться увеличение дозы и достижение более высоких минимальных уровней
Заместительная терапия при вторичных иммунодефицитах (как приведено в разделе «Показания»)
Рекомендуемая доза вводится одинаковыми интервалами (примерно один раз в неделю) для достижения кумулятивной (суммарной) ежемесячной дозы примерно 0,2–0,4 г/кг (1,2–2,4 мл/кг) Для каждой отдельной дозы может потребоваться другой анатомический участок (место)
Минимальные уровни IgG следует измерять и оценивать вместе с частотой инфекции При необходимости дозу следует корректировать, чтобы добиться оптимальной защиты от инфекций Может потребоваться увеличение дозы для пациентов с персистирующей инфекцией; Уменьшение дозы можно рассмотреть для пациентов, у которых отсутствует инфекция
Пациенты пожилого возраста
Поскольку доза определяется с учетом массы тела и корректируется в соответствии с лечением и диагнозом, считается, что доза для пациентов пожилого возраста не отличается от дозы для пациентов в возрасте от 18 до 65 лет В клиническом исследовании препарат Кутаквиг оценивали у 3 пациентов старше 65 лет Для достижения желаемых уровней сывороточного IgG не требовались специальные дозы
Способ применения и дозы
Применяется только для подкожного введения
В случае лечения дома проведения подкожных инъекций должен начинать и контролировать врач, имеющий опыт ведения пациентов, лечащихся дома Пациент и/или опекун следует проинформировать и научить, как пользоваться инфузионным устройством, как проводить процедуру введения, как проводить асептическую обработку, как вести журнал лечения, как распознавать и какие меры необходимо принимать в случае серьезных побочных реакций
Препарат Кутаквиг можно вводить в такие места как живот, бедро, плечо и боковая поверхность бедра
Скорость инфузии
Корректировка скорости инфузии и объема инфузии на место введения базируется на переносимости пациента
Рекомендуется использовать начальную скорость введения 15 мл/час/место Начиная с 7 инфузии, при хорошей переносимости (см раздел «Особенности применения») скорость инфузии можно постепенно увеличить до 25 мл/час/место Рекомендуемая скорость инфузии в час для всех мест введения вместе 30 мл/час для первых 6 инфузий, затем постепенно увеличить до 50 мл/час и при хорошей переносимости до – 80 мл/час
Одновременно можно использовать более одного инфузионного устройства
Объем инфузии на место введения
Количество вводимого в конкретное место препарата различно У новорожденных и детей для каждых 5–15 мл место введения можно менять У взрослых дозы свыше 30 мл можно делить согласно желанию пациента Нет ограничений на количество мест ввода Места ввода должны быть на расстоянии не менее 5 см друг от друга
Дети
Доза для детей (в возрасте 0–18 лет) не отличается от дозы для взрослых, поскольку доза при каждом показании вводится с учетом массы тела и корректируется в соответствии с ответом на лечение согласно показаниям для заместительной терапии
Препарат Кутаквиг оценивали у 38 пациентов детского возраста (26 детей от 2–12 лет и
12 детей от 12–16 лет с первичным иммунодефицитом) Для достижения желаемых уровней сывороточного IgG не требовались специальной дозы для пациентов детского возраста
Передозировка
Последствия передозировки неизвестны
Побочные реакции
Краткий обзор профиля безопасности
Иногда могут возникать нежелательные реакции, такие как озноб, головные боли, головокружение, лихорадка, рвота, аллергические реакции, тошнота, артралгия (боль в суставах), низкое артериальное давление и умеренная боль в пояснице
Редко иммуноглобулины человека нормальные могут вызвать внезапное снижение АД и в отдельных случаях – анафилактический шок, даже если у пациента не наблюдалось гиперчувствительности к предварительному введению препарата
Реакции в местах введения отек, болезненность, покраснение, уплотнение, чувство тепла в месте, кровоподтеки и сыпь могут возникать часто Частота таких реакций обычно уменьшается при продолжающемся лечении
Информацию о безопасности передачи микроорганизмов см в разделе «Особенности применения»
Список побочных реакций в виде таблицы
Данные о клинической безопасности препарата Кутаквиг базируются на опорном открытом проспективном многоцентровом исследовании III фазы с одной параллельной группой с участием пациентов с первичным иммунодефицитом, ранее проходили лечение вводимым внутривенно иммуноглобулином в течение по меньшей мере 6 месяцев Это исследование проводилось в Европе и Северной Америке
В данном исследовании безопасность препарата Кутаквиг оценивалась в 75 субъектах В общей сложности было введено 4462 инъекции препарата Кутаквиг
Представленная ниже таблица составлена в соответствии с классами систем органов (КСО) и сроками преимущественного использования медицинского словаря нормативно-правовой деятельности MedDRA
Частоту побочных реакций на одного пациента оценивали в соответствии с следующими условными обозначениями очень часто (≥ 1/10); часто (≥ 1/100 к по существующим данным)
В каждой группе частоты побочные действия представлены в порядке уменьшения их тяжести
Частота побочных реакций на одного пациента и одну инъекцию в клиническом исследовании с препаратом Кутаквиг
|
Класс системы органов (КСВ) MedDRA |
Побочная реакция |
Частота/инъекцию |
Частота/пациента |
|
Со стороны нервной системы |
Головная боль |
Редко |
Часто |
|
Со стороны желудочно-кишечного тракта |
Вздутие живота Боль в животе Рвота |
Редко Редко Редко |
Часто Часто Часто |
|
Со стороны скелетно-мышечной и соединительной ткани |
Миалгия (боль в мышцах) |
Редко |
Часто |
|
Общие нарушения и нарушения в месте введения |
Реакция в месте ввода Пирексия (гипертермия, лихорадка) |
Очень часто Редко |
Очень часто Часто |
|
исследование |
Положительная проба Кумбса Имеющийся свободный гемоглобин Сниженный гаптоглобин |
Редко Редко Редко |
Часто Часто Часто |
Нижеследующие побочные реакции были идентифицированы во время послерегистрационного применения препарата Кутаквиг Из-за того, что о таких побочных реакциях сообщало добровольно неопределенное количество пациентов, не всегда есть возможность надежно оценить их частоту или установить причинно-следственную связь с влиянием препарата
|
Класс системы органов (КСВ) MedDRA |
Побочная реакция (ПР) |
|
Со стороны нервной системы |
Головокружение |
|
Со стороны желудочно-кишечного тракта |
Тошнота |
|
Со стороны кожи и подкожных тканей |
Зуд |
|
Общие нарушения и нарушения в месте введения |
Усталость |
Сообщалось о таких дополнительных побочных реакциях во время подкожного применения иммуноглобулиновых препаратов после регистрации отек лица, тремор, бледность, бронхоспазм, диспное (затрудненное дыхание), кашель, диарея, крапивница, сыпь, гиперемия (приливы), ощущение жара, ощущение, астения (общая слабость), гриппоподобное заболевание, плохое самочувствие, боль в месте введения, ощущение сжатия в горле, асептический менингит, гипертензия и тромбоэмболические осложнения
Пациенты детского возраста
Следует ожидать, что частота, вид и тяжесть побочных реакций у детей такая же, как у взрослых
Сообщения о подозреваемых побочных реакциях
Сообщения о подозреваемых побочных реакциях после регистрации лекарственного препарата важны Это позволяет осуществлять непрерывный мониторинг баланса пользы и риска
Применение лекарственного препарата Врачей просят сообщать о любых подозреваемых побочных реакциях
Срок годности
3 года
После открытия флакона лекарственное средство следует использовать немедленно
Условия хранения
Хранить при температуре от 2 до 8 ˚С Не замораживать
Хранить флакон в наружной картонной упаковке для защиты от света
Хранить в недоступном для детей месте
В течение срока годности препарат можно хранить при комнатной температуре (ниже 25 С) до 6 месяцев без повторного хранения в холодильнике в этот период; препарат необходимо утилизировать, если его не использовали после хранения вне холодильника
Несовместимость
В связи с отсутствием исследований совместимости этот лекарственный препарат нельзя смешивать с другими лекарственными средствами
Упаковка
По 6 мл, 10 мл, 12 мл, 20 мл, 24 мл или 48 мл раствора для инъекций во флаконе По 1 флакону в картонной коробке
Категория отпуска
По рецепту
Производитель
1 Октафарма АБ
2 Октафарма Фармацевтика Продуктионсгес м б х
Адрес
1 Ларс Форсселлс Гата 23, Стокгольм, 11275, Швеция
2 Оберлаайер Штрассе 235, 1100 Вена, Австрия
Источником информации для описания является Государственный Реестр Лекарственных Средств Украины
Аналоги КУТАКВИГ
Киовиг — инструкция по применению
Синонимы, аналоги
Статьи
Регистрационный номер:
ЛП-004856
Торговое наименование:
Киовиг®
Группировочное наименование:
иммуноглобулин человека нормальный
Лекарственная форма:
раствор для инфузий
Состав:
1 мл содержит:
Действующее вещество: Иммуноглобулин человека нормальный 100 мг*
* белок плазмы человека, IgG не менее 98%
Вспомогательные вещества: глицин, вода для инъекций.
Один флакон содержит:
| Компонент | Количество | |||||
| Действующее вещество: | ||||||
| Иммуноглобулин человека нормальный (белок плазмы человека, IgG не менее 98%) | 1 г | 2,50 г | 5 г | 10 г | 20 г | 30 г |
| Вспомогательные вещества: | ||||||
| Глицин | 0,19 г | 0,47 г | 0,94 г | 1,88 г | 3,75 г | 5,63 г |
| Вода для инъекций до | 10 мл | 25 мл | 50 мл | 100 мл | 200 мл | 300 мл |
Описание:
прозрачный или слегка опалесцирующий, бесцветный или светло-желтый раствор.
Характеристика препарата
Распределение подклассов IgG:
IgG1 ≥56,9%
IgG2 ≥26,6%
IgG3 ≥3,4%
IgG4 ≥1,7%
Максимальное содержание иммуноглобулина A (IgA): 0,14 мг/мл.
Производится из плазмы человека.
Фармакотерапевтическая группа:
медицинский иммунобиологический препарат (МИБП)-глобулин
Код ATX:
J06BA02
Фармакологические свойства
Фармакодинамика
Препарат Киовиг® состоит преимущественно из иммуноглобулина G (IgG) с широким спектром функционально неповрежденных антител к инфекционным агентам. Обе функции Fc и Fab молекул IgG сохранены. Способность участков Fab связываться с антигенами была продемонстрирована биохимическими и биологическими методами. Функция Fc была протестирована путем активации комплемента и опосредованной Fc- рецептором активацией лейкоцитов. В препарате Киовиг® сохранено ингибирование индуцированной иммунным комплексом активации комплемента («очистка», противовоспалительное действие иммуноглобулина для внутривенного введения).
Препарат Киовиг® не приводит к неспецифической активации системы комплемента или прекалликреина.
Распределение подклассов IgG примерно соответствует их распределению в нормальной плазме человека. Оптимальные дозы препарата Киовиг® могут восстановить низкую концентрацию IgG до нормальных значений.
Механизм действия при применении согласно показаниям, за исключением заместительной терапии, не полностью объяснен, но включает в себя иммуномодулирующий эффект. Препарат повышает неспецифическую резистентность организма.
Дети
Различие эффекта иммуноглобулинов у детей по сравнению с взрослыми, согласно теоретическим представлениям и клиническим наблюдениям, отсутствует.
Фармакокинетика
Всасывание
Биодоступность нормального человеческого иммуноглобулина после внутривенного введения – немедленная и полная.
Распределение
Препарат относительно быстро распределяется в плазму крови и внеклеточную жидкость; через приблизительно 3-5 дней терапии достигается равновесие между внутри- и внесосудистыми компартментами.
Выведение
Фармакокинетические параметры для препарата Киовиг® были определены в двух клинических исследованиях при первичном иммунодефиците, проведенных в Европе и США. В этих исследованиях, в общей сложности, 83 пациента в возрасте минимум 6 лет получали препарат в дозах 300-600 мг/кг массы тела, каждые 21-28 дней в течение 6-12 месяцев. Медиана периода полувыведения IgG после введения препарата Киовиг® составила 32,5 дня. Данное значение может варьировать у различных пациентов, в частности, страдающих первичным иммунодефицитом. Полученные значения параметров фармакокинетики препарата суммированы в приведенной ниже таблице. Все параметры анализировались по отдельности в трех возрастных группах: у детей (в возрасте младше 12 лет; n = 5), подростков (в возрасте 13-17 лет; n = 10) и взрослых (в возрасте старше 18 лет; n = 64). Значения, полученные в этих исследованиях, сравнимы с отмечавшимися при использовании других препаратов иммуноглобулина человека.
Сводная информация о фармакокинетических параметрах препарата Киовиг®
| Показатель | Дети (≤12 лет) | Подростки (13-17 лет) |
Взрослые (≥18 лет) |
|||
| Медиана | 95% ДИ* | Медиана | 95% ДИ | Медиана | 95% ДИ | |
| Период полувыведения (дней) | 41,3 | 20,2-86,8 | 45,1 | 27,3-89,3 | 31,9 | 29,6-36,1 |
| Смин (мг/дл)/(мг/кг) (стабильный уровень) | 2,28 | 1,72-2,74 | 2,25 | 1,98-2,64 | 2,24 | 1,92-2,43 |
| Смакс (мг/дл)/(мг/кг) (пиковый уровень) | 4,44 | 3,30-4,90 | 4,43 | 3,78-5,16 | 4,50 | 3,99-4,78 |
| Восстановление in vivo (%) | 121 | 87-137 | 99 | 75-121 | 104 | 96-114 |
| Удельное восстановление (мг/дл) / (мг/кг) | 2,26 | 1,70-2,60 | 2,09 | 1,78-2,65 | 2,17 | 1,99-2,44 |
| AUC0-21 день (г · ч/дл) (площадь под кривой) | 1,49 | 1,34-1,81 | 1,67 | 1,45-2,19 | 1,62 | 1,50-1,78 |
*ДИ – доверительный интервал
IgG и IgG-комплексы разрушаются в клетках ретикулоэндотелиальной системы.
Доклинические данные по безопасности
Иммуноглобулины представляют собой нормальные компоненты организма человека. Безопасность препарата Киовиг® была продемонстрирована в нескольких доклинических исследованиях. Данные, полученные на основе стандартных доклинических исследований фармакологической безопасности и токсичности, не выявили риска для людей.
Проведение исследований токсичности препарата при его многократном введении, генотоксичности и репродуктивной токсичности у животных не представляет практической значимости ввиду образования антител к гетерогенным белкам. В процессе клинического применения препарата не было выявлено доказательств его канцерогенности. Экспериментальные исследования канцерогенности у различных видов животных не проводились.
Показания к применению
Заместительная терапия у взрослых, а также детей и подростков (в возрасте 0-18 лет) при следующих состояниях:
Первичные иммунодефициты с нарушением продукции антител, в том числе:
- врожденные агаммаглобулинемия и гипогаммаглобулинемия;
- общая вариабельная иммунная недостаточность;
- тяжелая комбинированная иммунная недостаточность;
- синдром Вискотта-Олдрича.
Вторичные иммунодефициты, в том числе:
- тяжелая форма вторичной гипогаммаглобулинемии и рецидивирующие бактериальные инфекции у больных хроническим лимфоидным лейкозом при неэффективности профилактической антибактериальной терапии;
- тяжелая форма вторичной гипогаммаглобулинемии и рецидивирующие бактериальные инфекции при неэффективности вакцинации пневмококковой вакциной при множественной миеломе;
- гипогаммаглобулинемия у пациентов, перенесших аллогенную трансплантацию гемопоэтических стволовых клеток;
- врожденный синдром приобретенного иммунодефицита (СПИД) у детей при наличии рецидивирующих инфекций.
Иммуномодулирующая терапия у взрослых, а также детей и подростков (в возрасте 0-18 лет) при следующих состояниях:
- Идиопатическая тромбоцитопеническая пурпура (ИТП) у детей или у взрослых при высоком риске кровотечений или перед хирургическими вмешательствами с целью коррекции количества тромбоцитов;
- Синдром Гийена-Барре;
- Болезнь Кавасаки;
- Мультифокальная моторная нейропатия (ММН).
Противопоказания
Гиперчувствительность к действующему веществу или к любому из вспомогательных веществ.
Повышенная чувствительность к гомологичным иммуноглобулинам, особенно в очень редких случаях дефицита иммуноглобулина A (IgA), когда у пациента присутствуют антитела к IgA.
Гиперпролинемия (очень редкое заболевание, которое затрагивает всего несколько семей в мире).
Применение при беременности и в период грудного вскармливания
Безопасность применения препарата Киовиг® в период беременности и период грудного вскармливания в контролируемых исследованиях не установлена.
Иммуноглобулины проникают через плаценту, особенно в третьем триместре беременности. Иммуноглобулины выделяются с грудным молоком, при этом антитела могут оказывать защитное действие у новорожденного. Таким образом, препарат Киовиг® в период беременности и период грудного вскармливания следует применять с осторожностью.
Большой опыт клинического применения иммуноглобулинов показывает, что возникновение отрицательного воздействия на течение беременности, плод или на новорожденного маловероятно.
Клинический опыт применения иммуноглобулинов свидетельствует об отсутствии нежелательного влияния на фертильность.
Способ применения и дозы
Доза и режим дозирования зависят от показания к применению. В случае заместительной терапии доза препарата может быть подобрана индивидуально для каждого пациента в зависимости от фармакокинетических параметров и клинического ответа. В качестве руководства рекомендуются следующие дозы препарата.
Заместительная терапия при первичных иммунодефицитах
Рекомендовано выбирать такой режим дозирования, при котором концентрация IgG повышается минимум до 5-6 г/л (определение содержания IgG проводят перед последующей инфузией). Равновесные концентрации достигаются через 3-6 месяцев после начала лечения. Рекомендуемая стартовая доза составляет от 0,4 до 0,8 г/кг массы тела, последующие дозы – не менее 0,2 г/кг массы тела каждые 3-4 недели.
Дозы препарата, необходимые для достижения концентрации IgG 5-6 г/л, составляют от 0,2 до 0,8 г/кг массы тела в месяц. Интервал между дозами, когда достигаются равновесные концентрации, варьируется от 3 до 4 недель. Следует измерять концентрации IgG для регулирования дозы и интервала введения.
Заместительная терапия при множественной миеломе с тяжелой формой вторичной гипогаммаглобулинемии и рецидивирующими бактериальными инфекциями при неэффективности вакцинации пневмококковой вакциной; хроническом лимфоидном лейкозе с тяжелой формой вторичной гипогаммаглобулинемии и рецидивирующими бактериальными инфекциями при неэффективности профилактической антибактериальной терапии; врожденном СПИД у детей при наличии рецидивирующих инфекций
Рекомендован режим дозирования от 0,2 до 0,4 г/кг массы тела каждые 3-4 недели.
Гипогаммаглобулинемия у пациентов с аллогенной трансплантацией гемопоэтических стволовых клеток
Дозы препарата, необходимые для поддержания концентрации IgG на уровне более 5 г/л) составляют от 0,2 до 0,4 г/кг массы тела каждые 3-4 недели.
Идиопатическая тромбоцитопеническая пурпура
В случае обострения назначают от 0,8 до 1 г/кг массы тела в первый день (возможно повторное однократное введение этой дозы еще один раз в последующие 3 дня) или по 0,4 г/кг массы тела ежедневно в течение 2-5 дней. В случае возникновения рецидива лечение может быть проведено повторно.
Синдром Гийена-Барре
0,4 г/кг массы тела в течение 5 дней. Имеется ограниченный опыт применения у детей.
Болезнь Кавасаки
От 1,6 до 2 г/кг массы тела отдельными равными дозами в течение 2-5 дней или одной дозой 2 г/кг массы тела однократно. Пациентам в качестве сопутствующей терапии необходимо назначать ацетилсалициловую кислоту.
Мультифокальная моторная нейропатия (ММН)*
Стартовая доза составляет 2 г/кг массы тела отдельными равными дозами в течение 2-5 дней подряд; последующие дозы в расчете 1 г/кг массы тела каждые 3 недели в течение 1-2 дней подряд.
Рекомендации по режимам дозирования приведены в следующей таблице.
| Показания | Дозы | Интервал между инъекциями |
| Заместительная терапия Первичные иммунодефициты |
Стартовая доза: 0,4-0,8 г/кг массы тела, далее: 0,2-0,8 г/кг массы тела | Однократно, каждые 3-4 недели до достижения концентрации IgG не менее 5-6 г/л. |
| Вторичные иммунодефициты | 0,2-0,4 г/кг массы тела | Однократно, каждые 3-4 недели до достижения концентрации IgG не менее 5- 6 г/л. |
| Дети с врожденным СПИД при наличии рецидивирующих инфекций | 0,2-0,4 г/кг массы тела | Однократно, каждые 3-4 недели. |
| Гипогаммаглобулинемия (<4 г/л) у пациентов с аллогенной трансплантацией гемопоэтических cmволовых клеток | 0,2-0,4 г/кг массы тела | Однократно, каждые 3-4 недели, для поддержания концентрации IgG более 5 г/л. |
| Применение в качестве иммуномодулятора Идиопатическая тромбоцитопеническая пурпура |
0,8-1 г/кг массы тела | В первый день; возможно повторное однократное введение в последующие 3 дня со дня первого введения. |
| или 0,4 г/кг массы тела |
Ежедневно в течение 2-5 дней. | |
| Синдром Гийена-Барре | 0,4 г/кг массы тела | Ежедневно в течение 5 дней. |
| Болезнь Кавасаки | 1,6-2 г/кг массы тела | Назначают равными дозами в течение 2-5 дней в сочетании с назначением ацетилсалициловой кислоты |
| или 2 г/кг массы тела |
Однократно в сочетании с назначением ацетилсалициловой кислоты. | |
| Мультифокальная моторная нейропатия (ММН)* | Стартовая доза: 2 г/кг массы тела; поддерживающая доза: 1 г/кг массы тела |
Назначают равными дозами в течение 2-5 дней. Каждые 3 недели в течение 1-2 дней подряд. |
*Лечение продолжительностью более 24 недель должно рассматриваться врачом и основываться на наблюдаемой реакции на применение препарата и оценке предполагаемого эффекта при долгосрочном лечении. Режим дозирования следует подбирать в соответствии с наблюдаемой клинической картиной заболевания.
Дети
Режим дозирования препарата у детей и подростков (0-18 лет) не отличается от такового у взрослых, поскольку доза для каждого показания определяется массой тела и корректируется в соответствии с клиническим исходом и приведенными выше условиями.
Способ применения
Препарат Киовиг® следует вводить только в виде внутривенных инфузий.
Препарат Киовиг® представляет собой раствор, готовый к применению. В случае хранения препарата в холодильнике перед введением температура раствора должна быть доведена до комнатной температуры. Для введения следует использовать инфузионную систему с воздушным клапаном и встроенным фильтром. Пробку флакона всегда следует прокалывать в центре в отмеченной зоне.
При необходимости разведения следует использовать 5% раствор декстрозы. Для получения раствора иммуноглобулина 50 мг/мл (5%) препарат Киовиг® раствор для инфузий 100 мг/мл (10%) следует развести равным объемом 5% раствора декстрозы. Техника работы в асептических условиях должна строго соблюдаться во время разведения препарата. Киовиг® нельзя смешивать с 0,9% раствором натрия хлорида. Раствор должен быть прозрачным или слегка опалесцирующим.
В случае помутнения раствора или присутствия в растворе механических включений препарат использованию не подлежит.
Неиспользованный препарат и расходные материалы следует утилизировать подходящим для этого способом.
Скорость введения
Препарат следует вводить первоначально со скоростью 0,3 мл/кг массы тела/ч (0,5 мг/кг массы тела/мин, приблизительно в течение 30 мин). В случае хорошей переносимости препарата скорость инфузии может быть постепенно увеличена до 4,8 мл/кг массы тела/ч (8 мг/кг массы тела/мин). У пациентов с первичными иммунодефицитами, которые хорошо переносили заместительную терапию препаратом, скорость инфузии может быть постепенно увеличена до максимального значения 7,2 мл/кг массы тела (12 кг/кгмассы тела/мин).
Побочное действие
За время проведения четырех клинических исследований были зарегистрированы серьезные нежелательные реакции, связанные с применением препарата, у 2-х из 160 пациентов (83 пациента 2-х исследований при первичном иммунодефиците, 23 пациента с идиопатической тромбоцитопенической пурпурой (ИТП) и 44 пациента, страдавших мультифокальной моторной нейропатией (ММН)): два эпизода асептического менингита у одного пациента, принимавшего участие в исследовании при первичном иммунодефиците, проводившемся в США, и один эпизод тромбоэмболии легочной артерии у пациента, принимавшего участие в клиническом исследовании при ММН. Большинство нежелательных явлений (НЯ), отмечавшиеся при использовании препарата по всем показаниям, имели легкую или среднюю степень тяжести.
В исследованиях препарата при первичном иммунодефиците общая частота нежелательных реакций на инфузию составила 0,27. Как и ожидалось, в исследовании при идиопатической тромбоцитопенической пурпуре частота нежелательных реакций на инфузию была выше вследствие использования значительно более высоких доз препарата (0,49); однако 87,5% этих нежелательных реакций были расценены как имевшие легкую степень тяжести. Общая частота нежелательных реакций на инфузию в исследовании при ММН составила 0,10.
Таблица нежелательных реакций
В приведенных ниже таблицах НЯ упорядочены по классам систем органов (КСО) и терминам предпочтительного употребления медицинского словаря терминологии регулятивной деятельности (MedDRA). В табл. 1 перечислены нежелательные явления, зарегистрированные в клинических исследованиях, в табл. 2 – зарегистрированные в процессе постмаркетингового применения препарата.
Использовались следующие категории частоты встречаемости: очень часто (>1/10); часто (≥1/100, но <1/10); нечасто (≥1/1000, но <1/100); редко (≥1/10 000, но <1/1000); очень редко (<1/10 000); не известна (невозможно оценить на основе имеющихся данных).
В рамках каждой категории частоты встречаемости нежелательные реакции перечислены в порядке снижения степени серьезности.
Таблица 1. Нежелательные реакции, зарегистрированные в рамках клинических исследований, по системам и органам
| Класс систем органов (КСО) MedDRA | Термин предпочтительного употребления MedDRA | Категория частоты встречаемости |
| Инфекции и инвазии | Бронхит, назофарингит | Часто |
| Хронический синусит, грибковая инфекция, инфекция верхних дыхательных путей, инфекция мочевыводящих путей, включая пиелонефрит, бактериальная инфекция мочевыводящих путей, асептический менингит | Нечасто | |
| Нарушения со стороны крови и лимфатической системы | Анемия, лимфаденопатия | Часто |
| Нарушения со стороны иммунной системы | Гиперчувствительность, ангионевротический отек, острая крапивница | Часто |
| Нарушения со стороны эндокринной системы | Нарушения со стороны щитовидной железы | Нечасто |
| Психические нарушения | Инсомния, тревожность | Часто |
| Раздражительность | Нечасто | |
| Нарушения со стороны нервной системы | Головная боль | Очень часто |
| Головокружение, мигрень, парестезия, гипестезия | Часто | |
| Амнезия, ощущение жжения, дизартрия, дисгевзия, нарушение равновесия, тремор | Нечасто | |
| Нарушения со стороны органа зрения | Конъюнктивит | Часто |
| Боль в глазу, отек конъюнктивы | Нечасто | |
| Нарушения со стороны органов слуха и равновесия | Головокружение | Часто |
| Наличие экссудата в среднем ухе | Нечасто | |
| Кардиологические нарушения | Тахикардия | Часто |
| Синусовая тахикардия | Нечасто | |
| Нарушения со стороны сосудистой системы | Гиперемия, гипертензия | Часто |
| Похолодание периферических отделов конечностей, флебит, приливы | Нечасто | |
| Нарушения со стороны дыхательной системы, грудной клетки и средостения | Кашель | Очень часто |
| Ринорея, бронхиальная астма, заложенность носа, боль в ротоглотке | Часто | |
| Диспноэ, отек ротоглотки | Нечасто | |
| Нарушения со стороны пищеварительной системы | Тошнота, рвота | Очень часто |
| Диарея, боли в животе | Часто | |
| Нарушения со стороны кожи и подкожных тканей | Кожный зуд, кожные высыпания, крапивница, эритематозные кожные высыпания, зудящие кожные высыпания | Часто |
| Холодный пот, дерматит, реакция фоточувствительности, ночная потливость, гипергидроз | Нечасто | |
| Нарушения со стороны костно-мышечной системы и соединительной ткани | Боли в конечности | Очень часто |
| Боль в спине, миалгия, мышечные спазмы, мышечная слабость | Часто | |
| Мышечные подергивания | Нечасто | |
| Нарушения со стороны мочевыделительной системы | Протеинурия | Нечасто |
| Системные нарушения и осложнения в месте введения | Боль в месте инфузии, боль в груди, отек в месте инфузии, озноб | Часто |
| Зуд в области применения, ощущение жара, флебит в месте инфузии, реакция, связанная с инфузией | Нечасто | |
| Изменения результатов лабораторных и инструментальных исследований | Повышение температуры тела, повышение артериального давления, снижение количества лейкоцитов, повышение активности аланинаминотрансферазы | Часто |
| Повышение концентрации холестерина в крови, повышение концентрации креатинина в крови, повышение концентрации мочевины в крови, снижение гематокрита, снижение количества эритроцитов, повышение частоты дыхательных движений | Нечасто | |
| Травмы, отравления и осложнения процедур | Ушиб | Часто |
| Прочие | Пирексия, утомляемость | Очень часто |
| Гриппоподобный синдром, дискомфорт в грудной клетке, чувство стеснения грудной клетки, боли в грудной клетке, астения, недомогание, периферические отеки | Часто |
Таблица 2. Нежелательные реакции, зарегистрированные в процессе постмаркетингового применения препарата, но системам и органам.
| Класс систем органов (КСО) MedDRA | Термин предпочтительного употребления MedDRA | Категория частоты встречаемости |
| Нарушения со стороны крови и лимфатической системы | Гемолиз | Неизвестно |
| Нарушения со стороны иммунной системы | Анафилактический шок, анафилактическая реакция | Неизвестно |
| Нарушения со стороны нервной системы | Транзиторная ишемическая атака, инсульт | Неизвестно |
| Нарушения со стороны сосудистой системы | Гипотензия, инфаркт миокарда, тромбоз глубоких вен | Неизвестно |
| Нарушения со стороны дыхательной системы, грудной клетки и средостения | Тромбоэмболия легочной артерии, отек легких | Неизвестно |
| Изменения результатов лабораторных и инструментальных исследований | Положительный результат прямой пробы Кумбса, снижение парциального давления кислорода в крови | Неизвестно |
| Травмы, отравления и осложнения процедур | Острое посттрансфузионное повреждение легких | Неизвестно |
Описание отдельных нежелательных реакций
Мышечные подергивания и слабость отмечались только у больных ММН.
Дети
Частота, виды и степень тяжести нежелательных явлений у детей были аналогичны таковым у взрослых.
Передозировка
Симптомы: передозировка препарата может приводить к гиперволемии и повышению вязкости крови, в особенности у пациентов группы риска, в частности пожилого возраста, а также страдающих патологией сердца и нарушением функции почек.
Дети
Дети в возрасте младше 5 лет могут быть особенно восприимчивы к перегрузке жидкостью. Поэтому необходим тщательный расчет дозы в данной популяции пациентов.
Дополнительно к этому, пациенты, страдающие болезнью Кавасаки, составляют группу особо высокого риска вследствие нарушения функции сердца, и скорость введения препарата у них следует строго контролировать.
Лечение: симптоматическое.
Взаимодействие с другими лекарственными средствами
Живые ослабленные вирусные вакцины
После терапии иммуноглобулинами эффективность живых вакцин против кори, эпидемического паротита, краснухи и ветряной оспы может быть снижена как минимум в течение 6 недель и до 3 месяцев. Следует соблюдать интервал в 3 месяца между назначением препарата и вакцинацией живыми аттенуированными вакцинами. В случае вакцинации против кори снижение эффективности вакцины может длиться до 1 года. Таким образом, у пациентов, привитых вакциной против кори, необходимо контролировать уровень антител.
Декстроза
Разведение препарата Киовиг® 5% раствором декстрозы может приводить к повышению концентрации глюкозы в крови.
Дети
Перечисленные ограничения применимы как к взрослым, так и к детям.
Особые указания
Несовместимость
Препараты Киовиг® нельзя смешивать с другими лекарственным средствами и с 0,9% раствором натрия хлорида. Тем не менее, разрешается разведение 5% раствором декстрозы. Установленные тяжелые нежелательные реакции могут быть связаны со скоростью введения препарата. Следует тщательно соблюдать скорость введения препарата, указанную в разделе «Способ применения и дозы». Необходимо проводить тщательный мониторинг пациентов и наблюдение на предмет возникновения любых симптомов на протяжении периода инфузии.
Некоторые нежелательные реакции могут наблюдаться более часто:
- в случае высокой скорости введения;
- у пациентов с гипогаммаглобулинемией или агаммаглобулинемией с недостаточностью IgA или без недостаточности IgA;
- у пациентов, которые получают терапию иммуноглобулином человека нормальным в первый раз, или в редких случаях, когда препарат иммуноглобулина человека нормального заменяют на Киовиг®, или при долгом перерыве после предыдущей инфузии.
Возможных осложнений можно избежать, если убедиться, что:
- у пациента не проявляется гиперчувствительность к иммуноглобулину человека нормальному при медленном введении препарата (0,3 мл/кг массы тела/ч);
- во время и после периода инфузии все симптомы, возникающие у пациентов, тщательно отслеживаются. В частности, пациентам, ранее не получавшим терапию иммуноглобулинами человека нормальными, а также переведенным с лечения другим препаратом иммуноглобулина для внутривенного введения, или при долгом интервале после предыдущей инфузии необходимо проводить мониторинг во время первой инфузии и в течение первого часа после первой инфузии для выявления потенциальных нежелательных явлений. Все другие пациенты должны находиться под наблюдением как минимум в течение 30 минут после применения препарата.
В случае развития нежелательного явления следует уменьшить скорость введения или прекратить введение препарата. Требуемое лечение зависит от характера и степени тяжести нежелательного явления.
В случае развития шока необходимо использовать стандартное лечение шоковых состояний.
Всем пациентам перед началом введения иммуноглобулина человека для внутривенного введения требуется соответствующая гидратация, мониторинг выделения мочи, мониторинг концентрации креатинина в сыворотке крови.
Гиперчувствительность
Истинные реакции гиперчувствительности встречаются редко. Они могут возникать в очень редких случаях при дефиците IgA с антителами к IgA.
Редко иммуноглобулин человека нормальный может быть причиной снижения артериального давления с развитием анафилактоидной реакции, даже у пациентов, которые ранее хорошо переносили терапию иммуноглобулином человека нормальным.
Хронические воспалительные демиелинизирующие полинейропатии
Имеется ограниченный опыт использования внутривенных иммуноглобулинов у пациентов с хроническими воспалительными демиелинизирующими полинейропатиями.
Гемолитическая анемия
Препараты иммуноглобулина человека для внутривенного введения могут содержать антитела против антигенов групп крови, которые могут действовать как гемолизины и связываться in vivo с эритроцитами, что может являться причиной положительного прямого антиглобулинового теста (проба Кумбса) и, редко, гемолиза. Гемолитическая анемия может развиваться после терапии препаратами иммуноглобулина человека для внутривенного введения в результате повышенной секвестрации эритроцитов. Зарегистрированы отдельные случаи развития нарушений функции почек и/или почечной недостаточности или синдрома диссеминированного внутрисосудистого свертывания, связанные с гемолизом.
Развитие гемолиза связано со следующими факторами риска: высокие дозы, независимо от введения в виде однократной дозы или отдельных доз в течение нескольких дней; а также группы крови А (II), В (III) и АВ (IV) в совокупности с сопутствующим наличием воспалительного процесса. При лечении пациентов с группами крови А (II), В (III) или АВ (IV) высокими дозами препарата по показаниям, отличным от ПИД, рекомендуется соблюдать повышенную осторожность. Имеются отдельные сообщения о случаях гемолиза у пациентов с ПИД, получающих заместительную терапию.
Необходимо проводить мониторинг клинических признаков и симптомов гемолиза у пациентов, получающих терапию препаратами иммуноглобулина человека для внутривенного введения. При возникновении признаков и/или симптомов гемолиза во время или после инфузии иммуноглобулина для внутривенного введения лечащий врач должен рассмотреть вопрос об отмене дальнейшего лечения (см. также раздел «Побочное действие»).
Острое посттрансфузионное повреждение легких (TRALI)
Были зарегистрированы случаи развития некардиогенного отека легких (острого посттрансфузионного повреждения легких – TRALI) у пациентов, получавших иммуноглобулин человека (в том числе, препарат Киовиг®).
Синдром асептического менингита (САМ)
При лечении препаратами иммуноглобулина для внутривенного введения были зарегистрированы случаи развития синдрома асептического менингита. После отмены иммуноглобулина для внутривенного введения в течение нескольких дней наступала ремиссия САМ без каких-либо последствий. Обычно этот синдром начинается в период от нескольких часов до 2 дней после лечения иммуноглобулином для внутривенного введения. При проведении анализа спинномозговой жидкости часто наблюдается плеоцитоз до нескольких тысяч клеток на мм³, как правило, за счет клеток гранулоцитарного ряда, а также повышенная концентрация белка, до нескольких сотен мг/дл.
САМ может развиваться чаще на фоне применения иммуноглобулина для внутривенного введения в высоких дозах (2 г/кг).
Тромбоэмболические осложнения
Имеются клинические данные о связи между применением иммуноглобулина человека для внутривенного введения и случаями возникновения тромбоэмболических осложнений, такими как инфаркт миокарда, острое нарушение мозгового кровообращения (включая инсульт), легочная тромбоэмболия и тромбоз глубоких вен, которые, предположительно, связаны с относительным увеличением вязкости крови при введении большого количества иммуноглобулинов. Необходимо соблюдать осторожность при назначении и проведении инфузий иммуноглобулинов для внутривенного введения пациентам с ожирением и пациентам с ранее установленными факторами риска развития тромботических осложнений, такими как преклонный возраст, артериальная гипертензия, сахарный диабет, тромбоэмболии или сердечно-сосудистое заболевание в анамнезе, случаи наследственной или приобретенной тромбофилии, продолжительный период нарушения подвижности, пациентам с тяжелой гиповолемией и пациентам с заболеваниями, при которых наблюдается увеличение вязкости крови.
Гиперпротеинемия, повышение вязкости сыворотки крови и последующая относительная псевдогипонатриемия могут отмечаться у пациентов, получающих человеческий иммуноглобулин. Это следует иметь в виду, поскольку начало терапии по поводу гипонатриемии (т.е. снижения содержания свободной воды в сыворотке крови) у этих пациентов может приводить к дополнительному повышению вязкости сыворотки крови и повышению риска тромбоэмболии.
У пациентов группы риска тромбоэмболий препараты человеческого иммуноглобулина должны вводиться с минимальной скоростью и использоваться в минимальной эффективной дозе.
Острая почечная недостаточность
Были выявлены случаи развития острой почечной недостаточности, острого некроза канальцев, нефропатии проксимальных канальцев и осмотического нефроза у пациентов, получавших терапию иммуноглобулином человека для внутривенного введен я. В большинстве случаев были определены факторы риска, такие как предшествующее наличие почечной недостаточности, сахарного диабета, гиповолемии, лишнего веса, сопутствующее лечение нефротоксичными препаратами или возраст старше 65 лет, сепсис или парапротеинемия.
В случае развития почечной недостаточности следует прервать терапию иммуноглобулином человека для внутривенного введения. Необходимо отметить, что среди сообщений о случаях развития нарушений функции почек или почечной недостаточности, развивающихся на фоне приема зарегистрированных препаратов иммуноглобулинов человека, доля препаратов, содержавших в качестве стабилизатора сахарозу, была непропорционально высока. Таким образом, для пациентов, которые находятся в группе риска, рекомендовано использование препаратов иммуноглобулина для внутривенного введения, которые не содержат сахарозу. Киовиг® не содержит в своем составе сахарозу или другие сахара.
Пациентам с риском развития острой почечной недостаточности или тромбоэмболических осложнений препараты иммуноглобулинов для внутривенного введения необходимо вводить с минимальной скоростью инфузии и в минимально возможной дозе.
Влияние на диагностические тесты
После введения иммуноглобулинов в крови пациента временно увеличивается число различных пассивно переданных антител, что может привести к ложноположительному результату в серологических тестах.
Пассивный перенос антител к антигенам эритроцитов, например, А, В и D, может привести к неверному результату в некоторых серологических тестах для определения антител к эритроцитам (например, проба Кумбса), при определении количества ретикулоцитов и в гаптоглобиновом тесте.
Информация по безопасности в отношении инфекционных агентов
Киовиг® производят из плазмы человека.
Стандартные меры по предотвращению передачи инфекций, возникающих в результате применения лекарственных препаратов, изготовленных из крови или плазмы человека, включают отбор доноров, проверку индивидуальных донаций и многоуровневую проверку пулов плазмы на наличие специфических маркеров инфекции, а также три стадии эффективной инактивации/или удаления вирусов при производстве препарата. Несмотря на это, при применении препаратов, изготовленных из крови или плазмы человека, нельзя полностью исключить возможность передачи инфекционных агентов. Это положение также применимо в отношении неизвестных или новых вирусов и других инфекционных агентов.
Меры, предпринимаемые для обеспечения противовирусной безопасности, считаются эффективными для вирусов, имеющих оболочку, таких как ВИЧ, вирусов гепатита В и С, а также для безоболочечных вирусов, таких как вирус гепатита А и парвовирус В19.
Получен обнадеживающий клинический опыт, указывающий на отсутствие передачи вируса гепатита А и парвовируса В19 с препаратами иммуноглобулина человека, и также предполагается, что наличие антител вносит значительный вклад в вирусную безопасность. Рекомендуется при каждом применении препарата Киовиг® регистрировать наименование и номер серии препарата, который вводится пациенту, для понимания связи между самочувствием пациента и использованной серии препарата.
Киовиг® нельзя применять после истечения срока годности, указанного на картонной пачке и этикетке флакона.
При необходимости разведения препарата до меньшей концентрации полученный раствор следует использовать непосредственно после приготовления.
Для получения раствора иммуноглобулина с концентрацией 50 мг/мл (5%) препарат Киовиг® с концентрацией 100 мг/мл (10%) должен быть разведен равным объемом раствора декстрозы.
В процессе разведения необходимо минимизировать риск микробной контаминации.
Перед введением препарат должен быть осмотрен на предмет наличия механических включений и изменения цвета. Раствор должен быть прозрачным или слегка опалесцирующим, бесцветным или бледно-желтым. Растворы, содержащие взвесь или осадок, не должны использоваться.
Любое количество неиспользованного препарата или отходы от его использования должны утилизироваться в соответствии с Российскими предписаниями.
Влияние на способность управлять транспортными средствами, механизмами
Некоторые нежелательные реакции, связанные с действием препарата, могут оказывать воздействие на способность управлять транспортным средством или движущимися механизмами. Для пациентов, у которых наблюдались нежелательные реакции при введении препарата Киовиг®, управление транспортным средством или движущимися механизмами возможно только после исчезновения симптомов нежелательных реакций.
Форма выпуска
Раствор для инфузий, 100 мг/мл, по 10, 25, 50, 100, 200 или 300 мл во флакон из гидролитического стекла типа I, укупоренный резиновой пробкой с алюминиевой обкаткой и пластмассовой крышкой. По одному флакону вместе с инструкцией по применению в картонной пачке.
Условия хранения
Хранить при температуре не выше 25° С.
Не замораживать!
Хранить флакон в картонной упаковке для защиты от действия света.
Хранить в недоступном для детей месте.
Срок годности
2 года.
Условия отпуска
Отпускается по рецепту.
Владелец регистрационного удостоверения:
Бакстер АГ, Австрия
Индустриештрассе 67, А-1221, Вена, Австрия
Производитель:
Бакстер С.А., Бельгия
Бульвар Рене Бранкуар 80, Лессины, 7860 Бельгия
Претензии потребителей, рекламации по качеству препарата Киовиг® и сообщения о развитии нежелательных реакций принимаются по адресу:
ЗАО Компания «Бакстер»
125171, г. Москва, Ленинградское шоссе, д. 16А, стр.1.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Киовиг® (Kiovig)
💊 Состав препарата Киовиг®
✅ Применение препарата Киовиг®
Описание активных компонентов препарата
Киовиг®
(Kiovig)
Приведенная научная информация является обобщающей и не может быть использована для принятия
решения о возможности применения конкретного лекарственного препарата.
Дата обновления: 2019.07.11
Владелец регистрационного удостоверения:
Код ATX:
J06BA02
(Иммуноглобулин нормальный человеческий для в/в введения)
Лекарственная форма
| Киовиг® |
Р-р д/инф. 100 мг/мл: 10 мг, 25 мл, 50 мл, 100 мл, 200 мл или 300 мл фл. рег. №: ЛП-004856 |
Форма выпуска, упаковка и состав
препарата Киовиг®
Раствор для инфузий прозрачный или слегка опалесцирующий, бесцветный или светло-желтый.
Вспомогательные вещества: глицин, вода д/и.
10 мл — флаконы (1) — пачки картонные.
25 мл — флаконы (1) — пачки картонные.
50 мл — флаконы (1) — пачки картонные.
100 мл — флаконы (1) — пачки картонные.
200 мл — флаконы (1) — пачки картонные.
300 мл — флаконы (1) — пачки картонные.
Фармакологическое действие
Человеческий Ig, восполняя дефицит антител, снижает риск развития инфекций у больных с первичным и вторичным иммунодефицитом.
Показания активных веществ препарата
Киовиг®
Врожденные иммунодефицитные состояния (врожденный полный или частичный иммунодефицит, вариационный иммунодефицит, тяжелые комбинированные иммунодефициты, синдром Вискотта-Олдрича); идиопатическая тромбоцитопеническая пурпура (особенно острые формы у детей).
Приобретенный иммунодефицит (хроническая лимфоцитарная лейкемия, СПИД у детей, трансплантация костного мозга и другие виды трансплантаций); синдром Кавасаки (в качестве дополнения к терапии ацетилсалициловой кислотой); профилактика и терапия инфекционных заболеваний.
Режим дозирования
Способ применения и режим дозирования конкретного препарата зависят от его формы выпуска и других факторов. Оптимальный режим дозирования определяет врач. Следует строго соблюдать соответствие используемой лекарственной формы конкретного препарата показаниям к применению и режиму дозирования.
В/в капельно.
Доза и схема введения зависят от показаний, возраста пациента и применяемой лекарственной формы.
Побочное действие
Головная боль, озноб, повышение температуры тела, тошнота, рвота, ломота в суставах, боль в спине, аллергические реакции.
Редко — снижение АД, в единичных случаях — анафилактический шок, симптомы асептического менингита (сильная головная боль, тошнота, рвота, повышение температуры тела, ригидность затылочных мышц, светочувствительность, нарушение сознания), усугубление почечной недостаточности у пациентов с нарушенной функцией почек.
Противопоказания к применению
Гиперчувствительность, дефицит IgA на фоне наличия у больного антител против IgA.
С осторожностью: беременность, период лактации.
Применение при беременности и кормлении грудью
С осторожностью применять при беременности и в период лактации (грудного вскармливания).
Применение у детей
Применение возможно по показаниям и в соответствии с режимом дозирования.
Особые указания
Временное повышение содержания введенных антител в крови пациента после введения Ig может обусловливать ложноположительные результаты серологических проб.
Не следует превышать рекомендуемую скорость введения препарата (вероятно развитие тяжелых побочных явлений). В течение всего периода инфузии и 20 мин после нее пациент должен находиться под врачебным контролем.
Лекарственное взаимодействие
Снижает активность ослабленных живых вакцин против кори, краснухи, эпидемического паротита, ветряной оспы (при введении в первые 2 нед после вакцинации против кори, паротита и краснухи прививки указанными вакцинами следует повторить не ранее чем через 3 мес).
Если вы хотите разместить ссылку на описание этого препарата — используйте данный код
Аналоги препарата
БиоГам Иммуноглобули…
(НПО МИКРОГЕН, Россия)
Габриглобин-IgG
(ИММУНО-ГЕМ, Россия)
Гамунекс®-С
(GRIFOLS THERAPEUTICS, США)
И.Г. Вена Н.И.В.
(KEDRION, Италия)
Имбиоглобулин
(НПО МИКРОГЕН, Россия)
Иммуновенин®
(НПО МИКРОГЕН, Россия)
Иммуноглобулин Сигар…
(СИГАРДИС РУС, Россия)
Иммуноглобулин Сигар…
(СИГАРДИС РУС, Россия)
Иммуноглобулин челов…
(НПО МИКРОГЕН, Россия)
Иммуноглобулин челов…
(НИЖЕГОРОДСКАЯ ОСПК ИМ. Н.Я.КЛИМОВОЙ ГУЗ, Россия)
Все аналоги
![]()
-
About us
-
Plasma
-
Products
-
Engagement
-
News
-
Careers and jobs at Octapharma
Homepage
Products
Our product portfolio
cutaquig®