Пертузумаб — это инновационный препарат для лечения HER2-положительного рака молочной железы (РМЖ), не отвечающего на терапию трастузумабом.
До того, как появилась таргетная терапия, HER2-позитивный РМЖ бы наиболее неблагоприятным вариантом опухоли в плане лечения и прогноза выживаемости. Ситуация кардинально изменилась с разработкой и внедрением в практику моноклональных антител против данного рецептора. Сегодня это считается одним из наиболее благоприятных и перспективных в плане лечения видов рака молочной железы. Однако не все пациенты с таким типом опухоли в полной мере отвечают на трастузумаб-содержащую терапию. У некоторых была первичная резистентность, когда опухоль была устойчива к проводимому лечению на этапе терапии первой линии. У других пациентов в течением времени развивалась вторичная резистентность . Поэтому поиски методов лечения продолжались, и одним из вариантов стало применение комбинации трастузумаба, пертузумаба и доцетаксела.
Фармакологическое действие пертузумаба
Действующее вещество пертузумаба представляет собой гуманизированное моноклональное антитело, которое связывается с внеклеточным доменом рецептора HER-2 и блокирует его соединение с другими рецепторами. Разберем как это работает.
Существует семейство рецепторов эпидермального фактора роста, которые запускают каскад реакций, обеспечивающих рост и размножение клеток. Если количество этих рецепторов значительно выше нормы, как это бывает в раковых клетках, они начинают бесконтрольно делиться и размножаться. В состав данного семейства входят 4 рецептора:
- HER-1.
- HER-2.
- HER-3.
- HER-4.
Все они, кроме HER-2, начинают работать после связывания с лигандами. Активность рецепторов проявляется только тогда, когда они связаны с другими рецепторами своего семейства. Такое состояние называется димеризация. Это могут быть гомодимеры, когда связывается два одинаковых рецептора, например HER-2/HER-2, или гетеродимеры, когда связываются неодинаковые рецепторы, например HER-2/HER-3.
Внеклеточная часть HER-2 имеет 4 части (субдомена), каждая из которых имеет свои функции. Трастузумаб блокирует действие четвертой части, а пертузумаб — второй, которая отвечает а димеризацию с другими рецепторами. Считается, что именно образование димера HER-2/HER-3 приводит к резистентности к трастузумабу. Для полноты противоопухолевого действия необходимо чтобы блокировались одновременно 2 субдомена, поэтому назначается одновременно 2 таргетных препарата — трастузумаб и пертузумаб.
Состав
Пертузумаб выпускается в виде концентрата, который подлежит растворению перед инфузионным введением.
Концентрат имеет вид прозрачной, опалесцирующей бесцветной жидкости. Допускается слегка коричневатый цвет. В 1 мл концентрата содержится 30 мг действующего вещества, в одном флаконе — 420 мг.
Препарат расфасован в бесцветные прозрачные стеклянные флаконы объемом 14 мл. Каждый флакон упакован в картонную коробку.
В качестве вспомогательных веществ применяются L-гистидин, уксусная кислота, сахароза, полисорбат и деионизированная вода.

Фармакологическое действие пертузумаба
Препарат оказывает противоопухолевое действие, блокируя гетеродимеризацию рецептора HER-2, тем самым подавляя инициацию процессов, необходимых для роста и размножения раковых клеток. Это в конечном итоге приводит к апоптозу.
Следует учитывать, что из-за высокой молекулярной массы основного действующего вещества, пертузумаб не может проникать через гематоэнцефалический барьер и поэтому неэффективен в отношении метастазов головного мозга.
Фармакокинетика
Проводилось изучение фармакокинетики пертузумаба только после его внутривенного введения. При этом использовались различные дозировки у больных с разными видами новообразования. Результаты показали, что фармакокинетических различий между начальными и последними стадиями рака груди нет.
Всасывание
Препарат вводится внутривенно. Изучение иных способов применения не проводилось.
Распределение
Пертузумаб распределяется в плазме и внеклеточной жидкости.
Метаболизм
Отдельного изучения особенностей метаболизма пертузумаба не проводилось. Предполагается, что как и все моноклональные антитела, он подвергается катаболическим процессам — окислению или распаду до более простых веществ.
Выведение
Период полувыведения 17,2-18,0 суток. Клиренс 0.235 л/сут.
Особые случаи
Не обнаружено изменения фармакокинетики пертузумаба в зависимости от половой принадлежности и этноса.
Наблюдались незначительные изменения клиренса пертузумаба, в зависимости от истинной массы тела (вес тела без учета жировой ткани) и уровня сывороточного альбумина. Но коррекции дозировки при изменении данных показателей не требовалось.
Фармакокинетические особенности пертузумаба у пожилых не исследовались. Но данные популяционных наблюдений свидетельствуют о том, что существенных различий нет.
Отдельных исследований, которые бы изучали особенности фармакокинетики пертузумаба у пациентов с почечной недостаточностью, не проводилось. Есть популяционные данные о том, что почечная недостаточность, в том числе тяжелой степени, не влияет на фармакокинетику препарата. Тем не менее, пока их нельзя назвать убедительными. Исследования при печеночной недостаточности не проводились.
Показания к применению
Пертузумаб применяют для лечения HER-2 позитивного РМЖ. Предварительно требуется проведение валидированных методов исследования на уровень экспрессии данного рецептора. К таким методам относят иммуногистохимический анализ (ИГХ) и гибридизацию in situ, в частности флуоресцентную гибридизацию (FISH). Критерием назначения таргетной терапии является 3 балла по ИГХ или амплификация гена по данным FISH.
Комбинация трастузумаба, пертузумаба и доцетаксела назначается в следующих случаях:
- Лечение РМЖ с отдаленными метастазами.
- Местно рецидивирующие опухоли РМЖ.
- Неоперабельный рак груди.
Условием назначение данной схемы является отсутствие в анамнезе ранее проводимого таргетного лечения и химиотерапии.
Комбинация пертузумаба, трастузумаба и цитостатиков (не таксанов) назначается при лечении РМЖ без отдаленных метастазов в следующих случаях:
- Предоперационная терапия раннего местно распространенного HER2+ РМЖ.
- Послеоперационное лечение раннего HER2+ РМЖ, когда опухоль превышает в размерах 2 см или уже дала регионарные метастазы.

Режим дозирования
Пертузумаб должен применяться как в условиях стационара, так и амбулаторно. Вводится он исключительно внутривенно капельно. Струйное или болюсное введение категорически запрещено.
Для приготовления готового препарата используется изотонический раствор NaCl. Применение для растворения декстрозы не рекомендуется, поскольку это приводит к химической и физической нестабильности.
Препарат разводят в асептических условиях. Учитывая то, что в его составе отсутствуют антимикробные консерванты, необходимо тщательно соблюдать условия стерильности. После разведения отбирают 14 мл приготовленного концентрированного раствора и вводят его в инфузионный пакет с изотоническим раствором NaCl (его объем должен составлять 250 мл). Допускается использование инфузионных пакетов на основе поливинилхлорида, полиэтилена или полиолефина.
Для равномерного перемешивания инфузионный пакет осторожно переворачивают несколько раз. После этого проверяют полученный раствор на предмет образования примесей или изменения цвета. Готовый раствор необходимо использовать немедленно. В крайних случаях допускается его хранение в течение суток в условиях холодильника. При этом необходимо удостовериться, что приготовление раствора было осуществлено по всем правилам асептики с соблюдением стерильности. Ответственность за качество раствора лежит на специалисте, который его приготовил.
Также пертузумаб нельзя смешивать с другими лекарственными препаратами. При комплексном применении необходимо последовательное введение лекарственных средств.
Во время первого введения дается нагрузочная доза, которая составляет 840 мг пертузумаба. Продолжительность инфузии должна составлять час. Далее препарат вводится каждые три недели в поддерживающей дозировке. Если последующее введение переносится хорошо, время инфузии можно сокращать. Максимально короткое время — 30 минут. После окончания процедуры пациентка остается под наблюдением в течение 30-60 минут.
Особенности применение в комплексной терапии
Трастузумаб
Таргетные препараты применяются последовательно. Последовательность может быть любая.
При первичном введении трастузумаба дается нагрузочная доза, которая составляет 8 мг/кг. Введение осуществляется в виде внутрикапельной инфузии в течение полутора часов. Поддерживающая доза составляет 6 мг/кг и применяется раз в три недели посредством 30-90-минутной капельницы. Время определяется переносимостью препарата у конкретной пациентки.
Вторым вариантом применения трастузумаба является введение фиксированной дозы, которая составляет 600 мг, независимот от массы тела пациентки. Режим введения такой же — раз в три недели в виде внутрикапельных 30-90 минутных инфузий. Нагрузочной дозы не требуется.
Лечение таксанами (доцетаксел)
Таксаны вводятся после таргетных препаратов. Дозировка при первом введении составляет 75 мг/м2. После этого инфузии проводят с интервалом 3 недели. Если все хорошо, дозировку увеличивают до 100 мг/м2 (если схема лечения не подразумевает применения карбоплатина).
Особенности терапии РМЖ 4 стадии
применяется таргетная терапия трастузумабом и пертузумабом совместно с доцетакселом. При этом доцетаксел используется в первых 6 циклах терапии, если нет признаков прогрессирования опухоли или явлений непереносимости. Необходимость его дальнейшего применения определяется врачом. Если доцетаксел отменяют, лечение продолжают только таргетными препаратами.
Особенности применения пертузумаба с паклитакселом
При комбинированном применении паклитаксела и пертузумаба, дозировка первого препарата должна составлять 80 мг/м2. Вводится он внутривенно инфузионно еженедельно.
Особенности адъювантной таргетной терапии пертузумабом и трастузумабом
Пациенты, которые получили комбинированное лечение на дооперационном этапе, могут продолжать принимать таргетную терапию в течение года после операции. Условием отмены лечения является потеря эффективности, о чем свидетельствует прогрессирование опухоли, или развитие тяжелых осложнений.
Если лечение проводилось в рамках послеоперационной терапии, ее также проводят в течение года (максимум 18 циклов), либо до прекращения эффекта или развития тяжелых осложнений.
Пропуск в применении препаратов
Если время между инфузиями составляет менее 6 недель, необходимо как можно скорее ввести препараты в поддерживающих дозировках.
Если прошло более 6 недель, терапию начинают заново с введения нагрузочных доз — 840 мг пертузумаба в виде часовой капельницы и 8 мг/кг трастузумаба в виде полуторачасовой капельницы. Затем лечение продолжают по поддерживающей схеме.
Коррекция дозы
Снижать дозировку таргетных препаратов не рекомендуется. При развитии токсических осложнений, лечение прекращают и возобновляют после нормализации состояния пациента. Если возникла необходимость в отмене трастузумаба, отменяют и пертузумаб.
Если на фоне химиотерапии развилось угнетение костного мозга, лечение продолжают, но необходимо контролировать развитие осложнений, которые связаны с миелосупрессией и нейтропенией в частности (инфекционные осложнения).
При развитии инфузионных реакций, уменьшают скорость введения препарата или на время прекращают процедуру.
При развитии анафилаксии, лечение немедленно прекращают и поднимают вопрос о его отмене.

Передозировка
Не проводилось изучения максимальной переносимой дозы и действия в дозировках, превышающих 25 мг/кг. В случаях подозрения на передозировки, необходимо установить контроль за состоянием пациента и, при необходимости, назначать симптоматическую терапию.
Побочные действия
Поскольку пертузумаб используется только в комбинации с другими препаратами, сложно установить влияние каждого отдельного препарата на развитие того или иного осложнения. Тем не менее, таковые имели место быть.
Наиболее частые осложнения, которые развивались более чем у 30% больных:
- Диарея.
- Выпадение волос.
- Тошнота.
- Упадок сил.
- Рвота.
- Снижение количества лейкоцитов.
- Тяжелая нейтропения с повышением температуры наблюдалась у 10% пациенток. Были даже зафиксированы летальные случаи.
Очень частые осложнения, которые встречались более чем в 1/10 случаев:
- Анемии.
- Инфузионные реакции.
- Нарушение аппетита, искажение вкусового восприятия.
- Бессонница.
- Головная боль, головокружение.
- Парестезии и периферические нейропатии.
- Слезоточивость.
- Кровотечения (в основном носовые).
- Кашель.
- Одышка.
- Стоматиты.
- Запоры.
- Сыпь.
- Кожный зуд.
- Поражение ногтей.
- Вагиниты.
- Риниты.
- Отеки.
- Боли различной локализации, в том числе мышцах и суставах.
Частые осложнения, которые развивались менее чем в 1/10 случаев и более чем в 1/100 случаев:
- Реакции гиперчувствительности.
- Нарушение функции левого желудочка.
- Паронихии.
- Инфекции ВДП (верхних дыхательных путей).
- Озноб.
Нечастые осложнения, которые развивались менее чем в 1/100 и более чем 1/1000 случаев:
- Застойная сердечная недостаточность.
- Развитие плеврального выпота.
- Развитие интерстициальной болезни легких.
Редкие осложнения, которые развивались менее чем в 1/1000 случаев:
- Анафилактический шок.
- Синдром высвобождения цитокинов.
- Синдром лизиса опухоли.
Противопоказания к применению
- Непереносимость схемы лечения.
- Необходимость отмены трастузумаба.
- ФВЛЖ ниже 50% (фракция выброса левого желудочка).
- Наличие в анамнезе застойной сердечной недостаточности.
- Артериальная гипертензия, которая не поддается контролю рекомендованными схемами лечения.
- Восстановление после инфаркта миокарда.
- Аритмии, требующие медикаментозной коррекции во время прима пертузумаба.
- Печеночная недостаточность.
- Беременность и кормление грудью.
- Возраст до совершеннолетия.
Лечение антрациклинами в кумулятивной дозировке превышающей 360 мг/м2.
Использование во время беременности и кормления грудью
Запрещено применение пертузумаба у беременных пациенток, поскольку есть риск развития порока почек, маловодия и даже гибели плода (данные получены во время исследований, проводимых на животных). Если необходимо лечение кормящей матери, рекомендуется отказаться от грудного вскармливания.
Также женщины, которые принимают данный препарат, должны использовать надежную контрацепцию на весь период лечения и в течение 6 месяцев после него.
Влияние препарата на фертильность человека не изучалось, но по данным исследований на животных, влияния на репродуктивную систему не наблюдалось.
Особые указания
Препарат применяется только после лабораторного подтверждения гиперэкспрессии HER-2.
В медицинских документах больного необходимо указывать торговое наименование препарата и его серийный номер.
Инфузия должна осуществляться при наличии доступа к набору оказания экстренной помощи и врача, имеющего опыт химиотерапевтического лечения.
Необходима осторожность при применении пертузумаба у пациентов с нарушением ФВЛЖ. До начала терапии им проводят исследование функции ФВЛЖ, а затем ее регулярный мониторинг. При ухудшении состояния, лечение отменяют до стабилизации.
Инфузионные реакции
В эту группу реакций было отнесено повышение температуры тела, озноб, упадок сил, рвота, головная боль. При развитии таких реакций, необходимо уменьшить скорость введения препарата и даже прервать его, если потребуется. Наблюдение за пациенткой устанавливается до устранения симптомов. При хорошей переносимости препаратов, пациентка должна находиться под наблюдением медперсонала на протяжении всей инфузии и в течение часа (в случае первого введения препарата) или получаса (при повторных кусах).
При развитии тяжелых инфузионных реакций рассматривается вопрос об отмене данного вида противоопухолевого лечения.
Реакции гиперчувствительности и анафилаксии
Наблюдались случаи летального исхода после анафилактической реакции на пертузумаб. В этой связи препарат вводится только при доступности аптечки экстренной помощи. При развитии анафилаксии, помощь необходимо начать оказывать немедленно. Дальнейшее лечение пертузумабом противопоказано.
Фебрильные нейтропении
Фебрильная нейтропения может развиться у пациентов, принимающих таргетную терапию совместно с доцетакселом. Наиболее высокие риски в течение первых 3 курсов. Чаще всего данное осложнение развивалось у больных на фоне диареи и мукозита. В этой связи рекомендуется симптоматическое лечение данных явлений.
Сроки годности и условия хранения
Пертузумаб должен храниться в условиях холодильника. Срок хранения — 2 года с момента изготовления. Утилизация неиспользованного препарата должна осуществляться согласно региональным нормативным актам. По мере возможности, пертузумаб не должен попадать в окружающую среду. Запрещается его утилизация посредством выливания в канализацию.
Фармакологическое действие
Противоопухолевое средство. Пертузумаб представляет собой рекомбинантные гуманизированные моноклональные антитела, которые избирательно взаимодействуют с отвечающим за димеризацию внеклеточным субдоменом II HER2 (рецептора эпидермального фактора роста человека 2 типа). Связывание пертузумаба с субдоменом II блокирует процесс лиганд-зависимой гетеродимеризации HER2 с другими белками семейства HER, включая EGFR (рецептор эпидермального фактора роста человека), HER3 (рецептора эпидермального фактора роста человека 3 типа) и HER4 (рецептора эпидермального фактора роста человека 4 типа). Таким образом, пертузумаб ингибирует лиганд-инициированную передачу внутриклеточных сигналов по двум основным сигнальным путям: путь митоген-активированной протеинкиназы (MAP) и путь фосфоинозитид-3-киназы (PI3K). Угнетение данных сигнальных путей способно привести к остановке роста клеток и апоптозу, соответственно. Кроме того, пертузумаб способствует активации антитело-зависимой клеточной цитотоксичности (АЗКЦ).
Молекулярная масса пертузумаба составляет около 148 кДа, и ожидается, что, как и другие моноклональные антитела, пертузумаб практически не проникает через ГЭБ.
Пертузумаб в виде моноагента ингибирует пролиферацию клеток опухоли человека. Показано усиление противоопухолевой активности пертузумаба на моделях ксенотрансплантатов с гиперэкспрессией HER2 при его применении в комбинации с трастузумабом.
Фармакокинетика
После в/в введения Vd в центральной камере составляет 3.07 л и приблизительно равен объему плазмы. Значения Vd в равновесном состоянии свидетельствуют о том, что распределение происходит только в плазме и внеклеточной жидкости.
Метаболизм пертузумаба не изучался. Как и другие антитела, пертузумаб преимущественно подвергается катаболизму.
T1/2 приблизительно равен 17.2 сут. Клиренс пертузумаба составляет приблизительно 0.239 л/сут и не зависел от дозы и показания.
Показания активного вещества
ПЕРТУЗУМАБ
Метастатический или местно-рецидивирующий, неоперабельный рак молочной железы с опухолевой гиперэкспрессией HER2: в комбинации с трастузумабом и доцетакселом при отсутствии ранее проводимого лечения или при прогрессировании заболевания после проведения адъювантной терапии.
Режим дозирования
До начала лечения пертузумабом необходимо провести тестирование на опухолевую экспрессию HER2. Обязательным критерием является 3+ балла по результатам иммуногистохимического анализа и/или степень амплификации ≥2.0 по результатам гибридизации in situ (ISH). Следует использовать точные и валидированные методы тестирования.
Вводят только в/в капельно.
Длительность инфузии при введении первой дозы составляет 60 мин. Если первая инфузия переносится хорошо, последующие можно проводить на протяжении 30-60 мин.
Применяют в комбинации с трастузумабом и доцетакселом.
Нагрузочная доза пертузумаба — 840 мг в виде в/в капельной инфузии в течение 60 мин. Далее через 3 недели и каждые 3 недели пертузумаб вводят в поддерживающей дозе 420 мг в виде в/в капельной инфузии в течение 30-60 мин.
При появлении признаков прогрессирования заболевания или неприемлемой токсичности лечение пертузумабом следует прекратить.
Снижение дозы пертузумаба не рекомендуется.
Пертузумаб следует отменить в случае, если отменено лечение трастузумабом. При отмене доцетаксела лечение пертузумабом и трастузумабом можно продолжать до прогрессирования заболевания или развития неприемлемой токсичности. Снижение дозы трастузумаба не рекомендуется.
Побочное действие
Со стороны системы кроветворения: очень часто — нейтропения, анемия, лейкопения, фебрильная нейтропения (в т.ч. с летальным исходом).
Со стороны иммунной системы: часто — гиперчувствительность.
Со стороны нервной системы: очень часто — периферическая невропатия, головная боль, головокружение, бессонница.
Со стороны органа зрения: очень часто — повышенное слезоотделение.
Со стороны сердечно-сосудистой системы: часто — нарушение функции левого желудочка, в т.ч. застойная сердечная недостаточность.
Со стороны дыхательной системы: очень часто — одышка; часто — плевральный выпот.
Со стороны пищеварительной системы: очень часто — снижение аппетита, извращение вкуса, диарея, тошнота, рвота, запор, стоматит.
Со стороны кожи и подкожных тканей: очень часто — алопеция, сыпь, патология ногтей, зуд, сухость кожи; часто — паронихий.
Со стороны костно-мышечной системы: очень часто — миалгия, артралгия.
Общие реакции: очень часто — повышенная утомляемость, астения, периферические отеки, воспаление слизистых оболочек различной локализации, повышение температуры тела, присоединение вторичных инфекций (инфекции верхних дыхательных путей, назофарингит).
Противопоказания к применению
Значения фракции выброса левого желудочка сердца (ФВЛЖ) до лечения ≤50%; застойная сердечная недостаточность в анамнезе; неконтролируемая артериальная гипертензия; недавно перенесенный инфаркт миокарда; серьезные нарушения сердечного ритма, требующие лекарственной терапии на момент необходимости применения пертузумаба, за исключением фибрилляции предсердий и пароксизмальной наджелудочковой тахикардии; предшествующее лечение антрациклинами с кумулятивной дозой доксорубицина или эквивалентного препарата >360 мг/м2; нарушения функции печени; беременность, период лактации (грудного вскармливания); возраст до 18 лет; повышенная чувствительность к пертузумабу.
Применение при беременности и кормлении грудью
Применение при беременности и в период лактации (грудного вскармливания) противопоказано.
Женщины, обладающие репродуктивным потенциалом, и женщины детородного возраста, являющиеся половыми партнерами пациентов, получающих пертузумаб, на фоне применения пертузумаба и в течение 6 месяцев после введения последней дозы, должны использовать эффективные методы контрацепции.
Применение у детей
Препарат противопоказан для применения у детей и подростков в возрасте до 18 лет
Особые указания
Требуется осторожность при снижении ФВЛЖ до уровня <50% на фоне предшествующей адъювантной терапии трастузумабом; предшествующем лечении антрациклинами или предшествующей лучевой терапии на область грудной клетки; состояниях, которые способны нарушать функцию левого желудочка; при нарушениях функции почек.
ФВЛЖ следует оценить перед применением пертузумаба и регулярно (например, каждые 3 месяца) определять на фоне лечения для того, чтобы убедиться, что ФВЛЖ находится в пределах нормальных значений, установленных в данном учреждении. Если ФВЛЖ составляет менее 40% или 40-45% при снижении на ≥10% от исходного уровня до лечения, применение пертузумаба и трастузумаба следует приостановить. Если после повторной оценки, проведенной в период приблизительно 3 недель, ФВЛЖ не улучшится или произойдет ее дальнейшее снижение, следует рассмотреть вопрос об отмене пертузумаба и трастузумаба, если только не будет решено, что преимущества их применения для конкретного пациента превосходят риск.
При введении пертузумаба следует тщательно наблюдать за пациентом на протяжении первой инфузии и в течение 60 мин после ее окончания, а также на протяжении последующих инфузий и в течение 30 мин после их окончания. При развитии клинически значимой реакции на инфузию следует замедлить скорость инфузии или прервать ее и провести соответствующие лечебные мероприятия. Тщательное наблюдение за пациентом и оценка его состояния рекомендуются до полного разрешения симптомов. У пациентов с тяжелыми инфузионными реакциями следует оценить необходимость полной отмены пертузумаба, с учетом степени тяжести наблюдавшейся реакции и характера ответа на лечение, назначенного в связи с нежелательной реакцией.
Фесго — инструкция по применению
Синонимы, аналоги
Статьи
Регистрационный номер
ЛП-007317
Торговое наименование
Фесго®
Международное непатентованное или группировочное наименование
Пертузумаб + Трастузумаб
Лекарственная форма
Раствор для подкожного введения
Состав
1 флакон (15 мл) с препаратом в дозировке 600 мг+600 мг/10 мл содержит:
действующие вещества: пертузумаб – 600 мг, трастузумаб – 600 мг;
вспомогательные вещества: рекомбинантная человеческая гиалуронидаза (rHuPH20), L-гистидин, L-гистидина гидрохлорида моногидрат, α,α-трегалозы дигидрат, сахароза, полисорбат 20, L-метионин, вода для инъекций до 10 мл.
1 флакон (20 мл) с препаратом в дозировке 1200 мг+600 мг/15 мл содержит:
действующие вещества: пертузумаб – 1200 мг, трастузумаб – 600 мг;
вспомогательные вещества: рекомбинантная человеческая гиалуронидаза (rHuPH20), L-гистидин, L-гистидина гидрохлорида моногидрат, α,α-трегалозы дигидрат, сахароза, полисорбат 20, L-метионин, вода для инъекций до 15 мл.
Описание
Прозрачная или опалесцирующая, от бесцветного до слегка коричневатого цвета жидкость.
Фармакотерапевтическая группа
Противоопухолевые средства, антитела моноклональные.
Код АТХ
L01XY02
Фармакологические свойства
Фармакодинамика
Механизм действия
Пертузумаб и трастузумаб представляют собой рекомбинантные гуманизированные моноклональные антитела IgG1κ (иммуноглобулин G, подкласс 1, легкая цепь κ), которые взаимодействуют с рецептором эпидермального фактора роста человека 2 типа (HER2, также известный как c-erbB-2), трансмембранного гликопротеина с собственной тирозинкиназной активностью. Пертузумаб и трастузумаб, не конкурируя между собой, связываются с определенными эпитопами HER2, субдоменами II и IV, и имеют комплементарные механизмы для нарушения передачи сигнала HER2. Применение пертузумаба и трастузумаба в комбинации приводит к усилению антипролиферативной активности in vitro и in vivo.
Кроме того, часть кристаллизующегося фрагмента (Fc) каркасов IgG1 пертузумаба и трастузумаба способствует мощной активации антитело-зависимой клеточной цитотоксичности (АЗКЦ). In vitro АЗКЦ пертузумаба и трастузумаба больше направлена на опухолевые клетки с гиперэкспрессией HER2, чем на опухолевые клетки без гиперэкспрессии HER2.
Иммуногенность
Как и при применении всех белковых лекарственных препаратов, у пациентов, получающих препарат Фесго®, может развиться иммунный ответ.
В исследовании FEDERICA частота возникновения антител к пертузумабу и трастузумабу, вызванных лечением, составила 6.1% (15/245) и 0.4% (1/245) соответственно у пациентов, получавших в/в пертузумаб и трастузумаб. Среди пациентов, у которых тест на антитела к пертузумабу был положительным, нейтрализующие пертузумаб антитела были обнаружены у двух пациентов.
Частота обнаружения антител к пертузумабу и трастузумабу в любой временной точке (включая начальный уровень) составила 10.3% (26/252) и 1.2% (3/252) соответственно у пациентов, получавших в/в пертузумаб в комбинации с трастузумабом. Среди таких пациентов нейтрализующие пертузумаб антитела были обнаружены у трех пациентов.
Частота возникновения антител к пертузумабу, трастузумабу и воргиалуронидазе альфа, вызванных лечением, составила 8.3% (20/241), 1.7% (4/241) и 3.8% (9/238) соответственно у пациентов, получавших терапию препаратом Фесго®. Среди таких пациентов нейтрализующие пертузумаб антитела были обнаружены у двух пациентов, а нейтрализующие трастузумаб антитела – у одного пациента.
Частота выявления антител к пертузумабу, трастузумабу и воргиалуронидазе альфа в любой временной точке (включая начальный уровень) составила 12.1% (30/248), 3.2% (8/248) и 9% (22/245) соответственно у пациентов, получавших терапию препаратом Фесго®. Среди таких пациентов нейтрализующие пертузумаб антитела были обнаружены у трех пациентов, нейтрализующие трастузумаб антитела – у одного пациента, нейтрализующие воргиалуронидазу альфа антитела – у одного пациента.
Клиническая значимость появления антител к пертузумабу, трастузумабу или воргиалуронидазе альфа после лечения препаратом Фесго® неизвестна.
Фармакокинетика
Экспозиции пертузумаба и трастузумаба после подкожного введения препарата Фесго® (нагрузочной дозы 1200 мг пертузумаба/600 мг трастузумаба с последующим введением 600 мг пертузумаба/600 мг трастузумаба каждые 3 недели) в опорном клиническом исследовании представлены в таблице 1. Фармакокинетические результаты для первичной конечной точки минимальной концентрации (Ctrough) пертузумаба в цикле 7 (до введения в цикле  показали отсутствие превосходства в/в пертузумаба (геометрическое среднее 72.4 мкг/мл) по сравнению с пертузумабом в составе препарата Фесго® (геометрическое среднее 88.7 мкг/мл). Соотношение геометрических средних для пертузумаба в составе препарата Фесго® и в/в пертузумаба составило 1.22 (90% доверительный интервал (ДИ): 1.14-1.31). Нижняя граница двустороннего 90% ДИ для соотношения геометрических средних пертузумаба в составе препарата Фесго® и в/в пертузумаба составила 1.14, то есть была выше заранее определенного предела 0.8.
показали отсутствие превосходства в/в пертузумаба (геометрическое среднее 72.4 мкг/мл) по сравнению с пертузумабом в составе препарата Фесго® (геометрическое среднее 88.7 мкг/мл). Соотношение геометрических средних для пертузумаба в составе препарата Фесго® и в/в пертузумаба составило 1.22 (90% доверительный интервал (ДИ): 1.14-1.31). Нижняя граница двустороннего 90% ДИ для соотношения геометрических средних пертузумаба в составе препарата Фесго® и в/в пертузумаба составила 1.14, то есть была выше заранее определенного предела 0.8.
Фармакокинетические результаты для вторичной конечной точки Ctrough трастузумаба в цикле 7 (до введения в цикле показали отсутствие превосходства в/в трастузумаба (геометрическое среднее 43.2 мкг/мл) по сравнению с трастузумабом в составе препарата Фесго® (геометрическое среднее 57.5 мкг/мл). Соотношение геометрических средних для трастузумаба в составе препарата Фесго® и в/в трастузумаба составило 1.33 (90% ДИ: 1.24-1.43).
Популяционная фармакокинетическая модель пертузумаба с линейным выведением из центральной камеры была спроектирована с использованием объединенных фармакокинетических данных по пертузумабу в составе препарата Фесго® и в/в пертузумабу. Эти данные были получены в опорном клиническом исследовании и использовались для описания наблюдаемых фармакокинетических концентраций пертузумаба после подкожного введения препарата Фесго® и в/в введения пертузумаба.
Популяционная фармакокинетическая модель с параллельным линейным и нелинейным выведением из центральной камеры была спроектирована с использованием объединенных фармакокинетических данных из исследования по сравнению трастузумаба для подкожного (п/к) введения и в/в трастузумаба для описания наблюдаемых фармакокинетических концентраций после в/в или п/к введения трастузумаба у пациентов с HER2-положительным ранним раком молочной железы.
Фармакокинетический анализ с использованием описанной популяционной фармакокинетической модели показал отсутствие влияния пертузумаба в составе препарата Фесго® на фармакокинетику трастузумаба в составе препарата Фесго®, поскольку фармакокинетика трастузумаба в составе препарата Фесго® и фармакокинетика п/к трастузумаба были сходными.
Спрогнозированные с помощью популяционной фармакокинетики экспозиции пертузумаба и трастузумаба представлены в таблице 1 ниже.
Таблица 1. Экспозиции пертузумаба и трастузумаба (средние 5й–95й перцентили) после подкожного введения препарата Фесго® или в/в введения пертузумаба или трастузумабаа
| Показатель | Пертузумаб в составе препарата Фесго® | В/в пертузумаб | Трастузумаб в составе препарата Фесго®б | В/в трастузумабб | |
| Ctrough (мкг/мл) | Цикл 5 | 85.1 (48.7–122.5) |
74.9 (47.8–99.8) |
27.7 (13.6–43.2) |
31.4 (21.1–50.9) |
| Цикл 7 | 88.9 (51.8–142.5) |
78.5 (41.3–114.9) |
57.5 (27.2–92.7) |
44.9 (29.7–76.2) |
|
| Максимальная концентрация (Cmax (мкг/мл)) | Цикл 5 | 106.5 (62.9–152.6) |
304.8 (191.1–409.7) |
44.6 (31.0–63.1) |
172.9 (133.7–238.9) |
| Цикл 7 | 149.5 (88.5–218.5) |
225.9 (158.5–301.8) |
117.3 (72.2–166.6) |
169.1 (130.6–238.9) |
|
| Площадь под кривой концентрация-время (AUC0 21 сутки (мкг/мл•сутки)) | Цикл 5 | 2306.9 (1388.4–3376.2) |
2519.7 (1898.4–3138.9) |
1023.8 (634.3–1442.6) |
1341.0 (1033.1–2029.0) |
| Цикл 7 | 2569.3 (1487.4–3786.1) |
2454.3 (1561.4–3346.1) |
1838.7 (1024.3–2715.5) |
1668.6 (1264.7–2576.9) |
Первую дозу препарата Фесго®, в/в пертузумаба и трастузумаба, вводили в цикле 5.
б Для симуляции фармакокинетики трастузумаба использовали популяционную фармакокинетическую модель согласно данным исследования по сравнению в/в трастузумаба и п/к трастузумаба.
Всасывание
Средняя Cmax в сыворотке крови и время до достижения максимальной концентрации (Tmax) для пертузумаба в составе препарата Фесго® составили 157 мкг/мл и 3.82 суток соответственно. На основании популяционного фармакокинетического анализа абсолютная биодоступность составила 0.712, а степень всасывания первого порядка (Ка) – 0.348 1/сутки.
Средняя Cmax в сыворотке крови и Tmax для трастузумаба в составе препарата Фесго® составили 114 мкг/мл и 3.84 суток соответственно. На основании популяционного фармакокинетического анализа абсолютная биодоступность составила 0.771, а Ка – 0.404 1/сутки.
Распределение
Согласно данным популяционного фармакокинетического анализа, объем распределения пертузумаба в составе препарата Фесго® в центральной камере (V¬с) составляет 2.77 л у среднестатистического пациента.
Согласно данным популяционного фармакокинетического анализа, объем распределения п/к трастузумаба в центральной камере (V¬с) составляет 2.91 л у среднестатистического пациента.
Метаболизм
Метаболизм препарата Фесго® специально не изучался. Антитела выводятся преимущественно путем катаболизма.
Выведение
На основании популяционного фармакокинетического анализа клиренс пертузумаба в составе препарата Фесго® составил 0.163 л/сутки, период полувыведения (t½) –приблизительно 24.3 суток.
На основании популяционного фармакокинетического анализа линейный клиренс п/к трастузумаба составил 0.111 л/сутки.
Установлено, что трастузумаб достигает концентраций <1 мкг/мл через 7 месяцев после введения последней дозы у ≥95% пациентов. Это составляет около 3% от популяционной рассчитанной минимальной концентрации в равновесном состоянии (Cmin,ss) или соответствует выведению ~97% препарата.
Фармакокинетика у особых групп пациентов
Пациенты детского возраста
Исследований по изучению фармакокинетики препарата Фесго® у пациентов детского возраста не проводилось.
Пациенты пожилого возраста
Исследований по изучению фармакокинетики препарата Фесго® у пациентов пожилого возраста не проводилось.
По результатам популяционного фармакокинетического анализа пертузумаба в составе препарата Фесго® и в/в пертузумаба возраст не оказывает значимого влияния на фармакокинетические параметры пертузумаба.
По результатам популяционного фармакокинетического анализа п/к или в/в трастузумаба возраст не влияет на распределение трастузумаба.
Пациенты с нарушением функции почек
Специального исследования фармакокинетики препарата Фесго® у пациентов с нарушением функции почек не проводилось.
На основании популяционного фармакокинетического анализа пертузумаба в составе препарата Фесго® и в/в пертузумаба было показано, что нарушение функции почек не оказывает влияния на экспозицию пертузумаба, однако данные по пациентам с нарушением функции почек тяжелой степени тяжести, включенные в популяционный фармакокинетичекий анализ, ограничены.
По данным популяционного фармакокинетического анализа п/к и в/в трастузумаба нарушение функции почек не влияет на распределение трастузумаба.
Пациенты с нарушением функции печени
Специального исследования фармакокинетики препарата Фесго® у пациентов с нарушением функции печени не проводилось.
Показания к применению
Ранний рак молочной железы
Препарат Фесго® показан к применению в комбинации с химиотерапией:
- в качестве неоадъювантной терапии у взрослых пациентов с HER2-положительным местно-распространенным, отечно-инфильтративным или ранним раком молочной железы c высоким риском рецидива;
- в качестве адъювантной терапии у взрослых пациентов с HER2-положительным ранним раком молочной железы с высоким риском рецидива.
Метастатический рак молочной железы
Препарат Фесго® показан к применению в комбинации с доцетакселом у взрослых пациентов с HER2-положительным метастатическим или местно-рецидивирующим, неоперабельным раком молочной железы при отсутствии ранее проводимой HER2 специфичной терапии или химиотерапии по поводу метастатического заболевания.
Противопоказания
Гиперчувствительность к пертузумабу, трастузумабу или к другим компонентам препарата в анамнезе.
Беременность и период грудного вскармливания.
Детский возраст до 18 лет (эффективность и безопасность применения не установлены).
Значения фракции выброса левого желудочка сердца (ФВЛЖ) до лечения <55% (ранний рак молочной железы) или <50% (метастатический рак молочной железы).
Застойная сердечная недостаточность в анамнезе.
Неконтролируемая артериальная гипертензия.
Недавно перенесенный инфаркт миокарда.
Серьезные нарушения сердечного ритма, требующие лекарственной терапии на момент назначения препарата Фесго®, за исключением фибрилляции предсердий и пароксизмальной наджелудочковой тахикардии.
Предшествующее лечение антрациклинами с кумулятивной дозой доксорубицина или эквивалентного препарата >360 мг/м2.
Нарушения функции печени (эффективность и безопасность применения не изучались).
С осторожностью
Снижение ФВЛЖ до уровня <50% на фоне предшествующей адъювантной терапии трастузумабом. Предшествующее лечение антрациклинами или предшествующая лучевая терапия на область грудной клетки; состояния, которые способны нарушать функцию левого желудочка; при нарушениях функции почек.
Применение при беременности и в период грудного вскармливания
Фертильность
Специальных исследований фертильности у животных для оценки эффектов препарата Фесго® не проводилось.
Специальных исследований фертильности у животных для оценки эффектов пертузумаба не проводилось. Нежелательного влияния на мужские и женские репродуктивные органы в ходе исследований токсичности повторных доз продолжительностью до 6 месяцев у яванских макак не отмечалось.
Репродуктивные исследования не выявили доказательств влияния на фертильность у самок яванских макак, получавших трастузумаб.
Контрацепция
Пациентки, обладающие детородным потенциалом, а также женщины, являющиеся половыми партнерами пациентов, которые получают препарат, должны использовать эффективные методы контрацепции на фоне применения препарата Фесго® и в течение 7 месяцев после введения последней дозы.
Беременность
Клинические исследования препарата Фесго® у беременных женщин не проводились. Внутривенное введение пертузумаба яванским макакам во время органогенеза приводило к маловодию, задержке развития почек и гибели эмбриона/плода. В рамках пострегистрационного применения трастузумаба беременными женщинами отмечались случаи нарушения роста и/или нарушения функции почек у плода в связи с маловодием, некоторые из которых приводили к фатальной гипоплазии легких у плода.
На основании вышеупомянутых исследований у животных и согласно пострегистрационным данным, препарат Фесго® при его применении беременной женщиной может оказывать повреждающее действие на плод. В случае наступления беременности необходимо предупредить женщину о возможности повреждающего действия на плод. Если беременная женщина получает терапию препаратом Фесго® или забеременеет во время лечения или в течение 7 месяцев после последней дозы препарата, то она должна находиться под тщательным наблюдением врачей разных специальностей.
Роды и родоразрешение
Безопасность применения препарата Фесго® во время родов и родоразрешения не установлена.
Грудное вскармливание
Поскольку IgG человека выделяется с грудным молоком, возможность всасывания и повреждающего действия на плод неизвестна. Таким образом, необходимо предупредить женщину о необходимости прекращения грудного вскармливания при применении препарата Фесго® и в течение 7 месяцев после приема последней дозы препарата.
Способ применения и дозы
Лечение препаратом Фесго® следует начинать только под руководством врача, имеющего опыт применения противоопухолевых препаратов.
Препарат Фесго® должен вводиться медицинскими специалистами, владеющими тактиками ведения пациентов с анафилаксией в условиях возможности незамедлительного начала полного спектра реанимационных мероприятий (см. раздел «Особые указания»).
Пациенты, которые в настоящее время получают пертузумаб и трастузумаб внутривенно, могут перейти на терапию препаратом Фесго®.
Переход от внутривенного введения пертузумаба и трастузумаба к подкожному введению препарата Фесго® (или наоборот) изучался в исследовании PHranceSCa (см. раздел «Побочное действие»).
Во избежание медицинских ошибок перед применением лекарственного препарата необходимо проверить этикетку на флаконе и убедиться, что используемый для приготовления и введения препарат представляет собой препарат Фесго®.
Режим дозирования
Для назначения терапии препаратом Фесго® пациенты должны иметь HER2-положительный статус опухоли, определяемый как 3+ балла по результатам иммуногистохимического анализа (IHC) и/или степень амплификации ≥2.0 по результатам гибридизации in situ (ISH). Следует использовать валидированные методы тестирования.
Тестирование должно быть выполнено в специализированной лаборатории, способной обеспечить валидацию процесса тестирования для обеспечения точных и воспроизводимых результатов. Подробные указания по проведению HER2-тестирования и интерпретации его результатов приведены в инструкциях по применению валидированных тест-систем, предназначенных для определения HER2 статуса.
Рекомендации по дозированию препарата Фесго® при метастатическом и раннем раке молочной железы представлены в таблице 2.
Таблица 2. Рекомендуемые способ применения и дозы препарата Фесго®
| Доза (независимо от массы тела) | Примерная длительность подкожной инъекции | Время наблюденияаб | |
| Нагрузочная доза | 1200 мг пертузумаба/ 600 мг трастузумаба | 8 минут | 30 минут |
| Поддерживающая доза (вводится через 3 недели после нагрузочной дозы и затем каждые 3 недели) | 600 мг пертузумаба/ 600 мг трастузумаба | 5 минут | 15 минут |
a За пациентами необходимо наблюдать на предмет возникновения реакций, связанных с введением, и реакций гиперчувствительности.
б Период наблюдения должен начинаться после введения препарата Фесго® и завершаться перед последующем введением химиотерапии.
Если пациент получил пертузумаб и трастузумаб внутривенно менее 6 недель назад, то рекомендуемая доза препарата Фесго® должна соответствовать поддерживающей дозе, т.е. 600 мг пертузумаба/600 мг трастузумаба каждые 3 недели. Если пациент получил пертузумаб и трастузумаб внутривенно ≥6 недель назад, то рекомендуемая доза препарата Фесго® должна соответствовать нагрузочной дозе, т.е. 1200 мг пертузумаба/600 мг трастузумаба, а затем поддерживающая доза 600 мг пертузумаба/600 мг трастузумаба через 3 недели после нагрузочной дозы и далее каждые 3 недели.
У пациентов, получающих таксаны, препарат Фесго® следует вводить до введения таксана.
При применении в комбинации с препаратом Фесго® рекомендуемая начальная доза доцетаксела составляет 75 мг/м2, которая может быть увеличена до 100 мг/м2 в зависимости от выбранного режима и переносимости начальной дозы. Также доцетаксел может применяться в дозе 100 мг/м2 каждые 3 недели в зависимости от выбранного режима.
Если пациент одновременно получает карбоплатин, то рекомендуемая доза доцетаксела составляет 75 мг/м2 в течение всей терапии (увеличение дозы не предусмотрено).
В случае применения в качестве адъювантной терапии рекомендуемая доза паклитаксела составляет 80 мг/м2 один раз в неделю в течение 12 циклов.
У пациентов, получающих лечение по схеме на основе антрациклинов, препарат Фесго® следует вводить после завершения полного курса антрациклинов (см. раздел «Особые указания»).
Метастатический рак молочной железы
Препарат Фесго® следует применять в комбинации с доцетакселом. Лечение препаратом Фесго® проводится до прогрессирования заболевания или до развития признаков неприемлемой токсичности, в том числе и в случае отмены терапии доцетакселом (см. раздел «Особые указания»).
Ранний рак молочной железы
В качестве неоадъювантной терапии препарат Фесго® следует вводить в течение 3-6 циклов в комбинации с химиотерапией в составе полной схемы лечения раннего рака молочной железы.
В качестве адъювантной терапии препарат Фесго® следует вводить до момента, когда общая продолжительность терапии составит 1 год (максимум 18 циклов), или до прогрессирования заболевания, или до развития признаков неприемлемой токсичности, в зависимости от того, что произойдет раньше, в составе полной схемы лечения раннего рака молочной железы вне зависимости от времени проведения операции.
Терапия должна включать стандартную химиотерапию на основе антрациклина и/или таксана. Введение препарата Фесго® следует начинать в день 1 первого цикла таксан-содержащей химиотерапии и продолжать, в том числе, и в случае отмены химиотерапии.
Пропуск в плановом введении
Если интервал между двумя последовательными введениями составил:
- <6 недель — следует как можно скорее ввести поддерживающую дозу препарата Фесго® (600 мг пертузумаба/600 мг трастузумаба), а затем продолжить применение препарата каждые 3 недели;
- ≥6 недель — следует повторно ввести нагрузочную дозу препарата Фесго® (1200 мг пертузумаба/600 мг трастузумаба), а затем вводить препарат Фесго® в поддерживающей дозе (600 мг пертузумаба/600 мг трастузумаба) каждые 3 недели.
Коррекция дозы
Не рекомендуется снижение дозы препарата Фесго®. Лечение препаратом Фесго® может быть прекращено по решению лечащего врача.
В период возникновения обратимой миелосупрессии, вызванной химиотерапией, лечение может быть продолжено при условии тщательного контроля осложнений, обусловленных нейтропенией.
Указания по модификации дозы доцетаксела и других препаратов для химиотерапии представлены в инструкциях по медицинскому применению указанных препаратов.
Дисфункция левого желудочка
При любых признаках и симптомах, указывающих на застойную сердечную недостаточность, следует приостановить прием препарата Фесго® не менее чем на 3 недели. При подтверждении симптомов сердечной недостаточности применение препарата Фесго® следует прекратить (см. раздел «Особые указания»).
Пациенты с метастатическим раком молочной железы
Пациенты должны иметь значение ФВЛЖ до начала лечения ≥50%. Применение препарата Фесго® следует приостановить не менее чем на 3 недели в случае:
- Снижение ФВЛЖ составляет ≤40%;
- ФВЛЖ 40-45%, при условии, что ФВЛЖ снизилась на ≥10% по отношению к значениям до начала терапии.
Применение препарата Фесго® может быть возобновлено, если ФВЛЖ восстановилась до >45% или до 40-45%, при условии, что ФВЛЖ снизилась на <10% по отношению к значениям до начала терапии.
Пациенты с ранним раком молочной железы
Пациенты должны иметь значение ФВЛЖ до начала лечения ≥55% (≥50% после завершения антрациклинового компонента химиотерапии, если он проводился).
В случае снижения ФВЛЖ до <50%, при условии, что ФВЛЖ снизилась на ≥10% по отношению к значениям до начала терапии, следует приостановить применение препарата Фесго® не менее чем на 3 недели.
Применение препарата Фесго® может быть возобновлено, если ФВЛЖ восстановилась до ≥50%, при условии, что ФВЛЖ снизилась на <10% по отношению к значениям до начала терапии.
Особые указания по дозированию
Пациенты пожилого возраста
В целом различий в эффективности препарата Фесго® у пациентов в возрасте ≥65 и <65 лет не наблюдалось. Коррекции дозы препарата Фесго® у пациентов в возрасте ≥65 лет не требуется. Имеются ограниченные данные по пациентам >75 лет.
Информацию по безопасности препарата Фесго® у пациентов пожилого возраста см. в разделе «Побочное действие».
Пациенты с нарушением функции почек
Коррекции дозы препарата Фесго® у пациентов с нарушением функции почек легкой или средней степени тяжести не требуется. Данные о фармакокинетике у пациентов с тяжелым нарушением функции почек ограничены, поэтому дать специальные указания по дозированию не представляется возможным (см. раздел «Фармакологические свойства)».
Пациенты с нарушением функции печени
Эффективность и безопасность препарата Фесго® у пациентов с нарушением функции печени не изучались. Маловероятно, что пациентам с нарушением функции печени потребуется коррекция дозы препарата Фесго®. Таким образом, специальная коррекция дозы не рекомендуется (см. раздел «Фармакологические свойства»).
Пациенты детского возраста
Эффективность и безопасность препарата Фесго® у детей и подростков в возрасте до 18 лет не установлены. Нет данных о применении препарата Фесго® в детской популяции для лечения рака молочной железы.
Способ применения
Препарат Фесго® предназначен для подкожного введения только в область бедра. Препарат Фесго® не предназначен для внутривенного введения.
Инъекции следует производить попеременно только в левое и правое бедро. Место новой инъекции должно отстоять от предыдущего минимум на 2.5 см, располагаться на здоровом участке кожи и не затрагивать участки покраснения, синяков, болезненности и уплотнений. Не следует разделять дозу между двумя шприцами или между двумя местами введения.
В ходе лечения препаратом Фесго® следует использовать другие места введения для подкожного введения других препаратов.
Нагрузочная и поддерживающая дозы должны вводиться в течение 8 и 5 минут соответственно.
Рекомендованный период наблюдения для нагрузочной дозы составляет 30 минут, для поддерживающей – 15 минут для отслеживания реакций, связанных с введением (см. разделы «Побочное действие» и «Особые указания»).
Реакции, связанные с введением
Следует уменьшить скорость инъекции или на время прекратить введение препарата при развитии симптомов реакции, связанной с введением (см. разделы «Побочное действие» и «Особые указания»). Кислородотерапия, бета-агонисты, антигистаминные препараты, быстрое внутривенное введение жидкостей и жаропонижающие средства могут быть использованы с целью облегчения системных симптомов.
Реакции гиперчувствительности/анафилаксии
Следует немедленно прервать введение препарата и полностью отменить терапию в случае развития реакции 4 степени тяжести по классификации NCI-CTCAE (анафилаксии), бронхоспазма или острого респираторного дистресс-синдрома (см. разделы «Побочное действие» и «Особые указания»).
Обращение с препаратом
1200 мг пертузумаба/600 мг трастузумаба и 600 мг пертузумаба/600 мг трастузумаба в лекарственной форме «раствор для подкожного введения» являются готовыми к использованию растворами для инъекций, которые не нужно растворять в других лекарственных препаратах или смешивать.
Перед введением раствор препарата Фесго® следует проверить (визуально) на предмет отсутствия механических примесей и изменения окраски. Не встряхивать.
Флакон с препаратом используется только однократно. Подготовка препарата к введению должна проводиться в асептических условиях.
Так как раствор препарата Фесго® не содержит антимикробного консерванта, с микробиологической точки зрения препарат должен быть использован сразу после его переноса из флакона в шприц.
После набора раствора в шприц рекомендуется заменить иглу-переходник на закрывающий колпачок шприца во избежание высыхания раствора в игле и снижения качества лекарственного препарата. Следует наклеить на шприц отрывную часть этикетки флакона. Игла для подкожного введения должна быть присоединена к шприцу непосредственно перед введением с корректировкой объема раствора до 10 мл (600 мг пертузумаба/600 мг трастузумаба) или 15 мл (1200 мг пертузумаба/600 мг трастузумаба).
Не отмечалось признаков несовместимости препарата Фесго® с полипропиленом, поликарбонатом, полиуретаном, полиэтиленом, поливинилхлоридом и фторированным этилен-полипропиленом.
Побочное действие
Резюме профиля безопасности
Наиболее частыми нежелательными реакциями (≥30%), которые отмечались у пациентов, получавших препарат Фесго® или в/в пертузумаб в комбинации с трастузумабом и химиотерапией, были алопеция, диарея, тошнота, анемия, астения и артралгия.
Наиболее частыми серьезными нежелательными явлениями (≥1%), отмечавшимися у пациентов, получавших препарат Фесго® или в/в пертузумаб в комбинации с трастузумабом, были фебрильная нейтропения, сердечная недостаточность, повышение температуры тела, нейтропения, нейтропенический сепсис, снижение числа нейтрофилов и пневмония.
Профиль безопасности препарата Фесго® в целом соответствовал известному профилю безопасности в/в пертузумаба в комбинации с трастузумабом; дополнительно отмечались нежелательные реакции, связанные с введением (14.9% по сравнению с 0.4%).
Табличное резюме нежелательных реакций
Безопасность пертузумаба в комбинации с трастузумабом была оценена у 3344 пациентов с HER2-положительным раком молочной железы в опорных исследованиях CLEOPATRA, NEOSPHERE, TRYPHAENA и APHINITY. В целом показатели безопасности были схожими во всех исследованиях, хотя частота и наиболее частые нежелательные реакции различались в зависимости от того, назначался ли пертузумаб в комбинации с трастузумабом с или без сопутствующей терапии противоопухолевыми препаратами.
Безопасность препарата Фесго® была оценена у 248 пациентов с HER2-положительным ранним раком молочной железы в опорном клиническом исследовании FEDERICA.
В таблице 3 представлены нежелательные реакции, о которых сообщалось в связи с применением в/в пертузумаба в комбинации с трастузумабом и химиотерапией в указанных ниже опорных клинических исследованиях (n=3344) и при пострегистрационном применении. Также в таблицу 3 включены нежелательные реакции, специфичные для пути введения препарата Фесго®, о которых сообщалось в исследовании FEDERICA.
- CLEOPATRA, в котором пертузумаб назначался в комбинации с трастузумабом и доцетакселом пациентам с метастатическим раком молочной железы (n=453).
- NEOSPHERE (n=309) и TRYPHAENA (n=218), в которых в качестве неоадъювантной терапии применялся пертузумаб в комбинации с трастузумабом и химиотерапией у пациентов с местно-распространенным, отечно-инфильтративным или ранним раком молочной железы.
- APHINITY, в котором в качестве адъювантной терапии применялся пертузумаб в комбинации с трастузумабом и химиотерапией на основе таксана в сочетании с антрациклин-содержащим режимом или без него у пациентов с ранним раком молочной железы (n=2364).
- FEDERICA, в котором препарат Фесго® (n=248) или в/в пертузумаб и трастузумаб (n=252) применялись в комбинации с химиотерапией у пациентов с ранним раком молочной железы.
Поскольку пертузумаб применяется в комбинации с трастузумабом и химиотерапией, трудно установить причинно-следственную связь нежелательной реакции с конкретным лекарственным препаратом.
Ниже представлены нежелательные реакции, которые сгруппированы в соответствии с классами систем органов и категориями частоты медицинского словаря для нормативно-правовой деятельности MedDRA:
- очень часто (≥1/10);
- часто (≥1/100 и <1/10);
- нечасто (≥1/1000 и <1/100);
- редко (≥1/10000 и <1/1000);
- очень редко (<1/10000);
- неизвестно (частота не может быть определена на основании имеющихся данных).
В рамках каждой категории частоты нежелательные реакции представлены в порядке снижения серьезности.
Таблица 3. Резюме нежелательных реакций у пациентов, получавших терапию пертузумабом, трастузумабом в опорных1 клинических исследованиях и при пострегистрационном применении7
| Класс систем органов | Очень часто | Часто | Нечасто | Редко |
| Инфекционные и паразитарные заболевания | Назофарингит | Паронихия Инфекции верхних дыхательных путей |
||
| Нарушения со стороны крови и лимфатической системы | Фебрильная нейтропения2 Нейтропения Лейкопения Анемия |
|||
| Нарушения со стороны иммунной системы | Инфузионные реакции2,5 | Реакции гиперчувствительности2,4 Лекарственная гиперчувствительность4 |
Анафилактические реакции4 | Синдром высвобождения цитокинов5 |
| Нарушения со стороны обмена веществ и питания | Снижение аппетита | Синдром лизиса опухоли7 | ||
| Нарушения психики | Бессонница | |||
| Нарушения со стороны нервной системы | Периферическая нейропатия Головная боль Дисгевзия Периферическая сенсорная нейропатия Головокружение Парестезия |
|||
| Нарушения со стороны органа зрения | Повышенное слезоотделение | |||
| Нарушения со стороны сердца | Дисфункция левого желудочка3 | Застойная сердечная недостаточность3 | ||
| Нарушения со стороны сосудов | Приливы | |||
| Нарушения со стороны дыхательной системы, органов грудной клетки и средостения | Кашель Носовое кровотечение Одышка |
Интерстициальная болезнь легких Плевральный выпот |
||
| Нарушения со стороны желудочно-кишечного тракта | Диарея Рвота Стоматит Тошнота Запор Диспепсия Боль в животе |
|||
| Нарушения со стороны кожи и подкожных тканей | Алопеция Сыпь Патология ногтей Зуд Сухость кожи |
|||
| Нарушения со стороны скелетно-мышечной и соединительной ткани | Миалгия Артралгия Боль в конечностях |
|||
| Общие расстройства и нарушения в месте введения | Воспаление слизистых оболочек Периферические отеки Повышение температуры тела Повышенная утомляемость Астения Реакции в месте инъекции6 |
Озноб Боль Отеки |
1 В таблице 3 представлены объединенные данные за общий период терапии в исследовании CLEOPATRA (дата среза данных 11 февраля 2014 года; среднее число циклов пертузумаба составило 24); за период неоадъювантной терапии в исследованиях NEOSPHERE (среднее число циклов пертузумаба составило 4, по всем группам лечения) и TRYPHAENA (среднее число циклов пертузумаба составило 3-6 по группам лечения); а также за период лечения в исследовании APHINITY (среднее число циклов пертузумаба составило 18). Также в таблице 3 приведены данные о нежелательных реакциях, характерных для способа введения препарата Фесго®, о которых сообщалось в исследовании FEDERICA (среднее число циклов препарата Фесго® составило 7).
2 В том числе нежелательные реакции с летальным исходом.
3 Для общего периода терапии в 4 исследованиях (CLEOPATRA, NEOSPHERE, TRYPHAENA, APHINITY). Частота дисфункции левого желудочка и застойной сердечной недостаточности отражает предпочтительные термины MedDRA, отмечавшиеся в отдельных исследованиях.
4 Реакции гиперчувствительности/анафилаксии основаны на группе терминов.
5 Инфузионные реакции включают ряд различных терминов в рамках временного интервала, определяемые как любые системные явления, зарегистрированные как гиперчувствительность, анафилактическая реакция, острая инфузионная реакция или синдром высвобождения цитокинов, возникающие во время инфузии или в течение 24 часов после инфузии.
6 Наблюдались только при применении препарата Фесго®.
7 Нежелательные реакции, отмечавшиеся при пострегистрационном применении.
Описание отдельных нежелательных реакций
Дисфункция левого желудочка
Препарат Фесго® в комбинации с химиотерапией
В опорном клиническом исследовании FEDERICA частота симптоматической сердечной недостаточности (III или IV класса по классификации Нью-Йоркской кардиологической ассоциации (NYHA)) со снижением ФВЛЖ на ≥10% от начального уровня и до уровня <50% составила 1.2% при применении препарата Фесго® и 0.8% при применении в/в пертузумаба и трастузумаба. Ни у одного из пациентов с симптоматической сердечной недостаточностью, получавших препарат Фесго®, явление не разрешилось (на момент среза данных); при этом у одного пациента терапия препаратом Фесго® была прекращена в связи с симптоматической сердечной недостаточностью. Частота бессимптомного снижения ФВЛЖ на ≥10% от начального уровня и до уровня <50% или со слабо выраженными симптомами (класс II по классификации NYHA) (подтверждено вторичным анализом ФВЛЖ) составила 0.8% при применении препарата Фесго® и 4% при применении в/в пертузумаба и трастузумаба; при этом у пациента, получавшего препарат Фесго®, явление разрешилось (на момент среза данных), у двух пациентов терапия препаратом Фесго® была прекращена (см. разделы «Способ применения и дозы» и «Особые указания»).
Пертузумаб в/в в комбинации с трастузумабом и химиотерапией
В опорном клиническом исследовании CLEOPATRA частота дисфункции левого желудочка во время лечения была выше в группе плацебо, чем в группе пертузумаба (8.6% и 6.6% соответственно). Частота симптоматической дисфункции левого желудочка была также ниже в группе пертузумаба (1.8% в группе плацебо по сравнению с 1.5% в группе пертузумаба) (см. раздел «Особые указания»).
На фоне неоадъювантной терапии по данным исследования NEOSPHERE, в котором пациенты получили 4 цикла пертузумаба в качестве неоадъювантной терапии, частота дисфункции левого желудочка (в течение всего периода лечения) была выше в группе пертузумаба, трастузумаба и доцетаксела (7.5%) по сравнению с группой трастузумаба и доцетаксела (1.9%). В группе пертузумаба и трастузумаба зарегистрирован случай развития симптоматической дисфункции левого желудочка.
На фоне неоадъювантной терапии по данным исследования TRYPHAENA частота дисфункции левого желудочка (в течение всего периода лечения) составила 8.3% в группе пациентов, получавших лечение пертузумабом, трастузумабом и FEC (5-фторурацил, эпирубицин, циклофосфамид), с последующим применением пертузумаба, трастузумаба и доцетаксела; 9.3% в группе пациентов, получавших лечение пертузумабом, трастузумабом и доцетакселом после терапии FEC; 6.6% в группе пациентов, получавших лечение пертузумабом в комбинации с TCH (доцетаксел, карбоплатин, трастузумаб). Частота симптоматической дисфункции левого желудочка (застойная сердечная недостаточность) составила 1.3% в группе пациентов, получавших лечение пертузумабом, трастузумабом и доцетакселом после терапии FEC (исключая пациента, у которого развилась симптоматическая дисфункция левого желудочка во время терапии FEC до начала терапии пертузумабом, трастузумабом и доцетакселом) и также 1.3% в группе пациентов, получавших лечение пертузумабом в комбинации с TCH. У пациентов, получавших пертузумаб, трастузумаб и FEC с последующим применением пертузумаба, трастузумаба и доцетаксела случаев симптоматической дисфункции левого желудочка не отмечалось.
В исследовании BERENICE на фоне неоадъювантной терапии пертузумабом в комбинации с трастузумабом и паклитакселом после предшествующей химиотерапии доксорубицином и циклофосфамидом с сокращенными интервалами частота симптоматической дисфункции левого желудочка III/IV класса по классификации NYHA (застойной сердечной недостаточности по классификации NCI-CTCAE, версия 4) составила 1.5%. Ни у одного из пациентов (0%) не развилась симптоматическая дисфункция левого желудочка в группе пациентов, получавших FEC с последующим применением пертузумаба в комбинации с трастузумабом и доцетакселом.
Частота бессимптомной дисфункции левого желудочка (предпочтительный термин – снижение фракции выброса по классификации NCI-CTCAE, версия 4) составила 7% у пациентов, получавших химиотерапию доксорубицином и циклофосфамидом с сокращенными интервалами, а затем пертузумаб в комбинации с трастузумабом и паклитакселом, и 3.5% у пациентов, получавших терапию пертузумабом, трастузумабом и доцетакселом, которой предшествовала терапия FEC.
В исследовании APHINITY частота симптоматической сердечной недостаточности (III или IV класса по классификации NYHA) со снижением ФВЛЖ на ≥10% от начального уровня и до уровня <50% была <1%: 0.6% у пациентов, получавших пертузумаб (у 46.7% явление разрешилось) и 0.3% у пациентов, получавших плацебо (у 57.1% явление разрешилось). Считалось, что явление разрешилось, если 2 последовательных измерения ФВЛЖ были с результатом >50% на момент даты среза данных.
Большинство явлений отмечалось у пациентов, получавших антрациклины.
Частота бессимптомного снижения ФВЛЖ на ≥10% от начального уровня и до уровня <50% или со слабо выраженными симптомами (класс II по классификации NYHA) составила 2.7% у пациентов, получавших пертузумаб (у 79.7% явление разрешилось на момент даты среза данных) и 2.8% у пациентов, получавших плацебо (у 80.6% явление разрешилось на момент даты среза данных).
Реакции, связанные с введением/инфузионные реакции
Препарат Фесго® в комбинации с химиотерапией
В опорном клиническом исследовании FEDERICA любые системные реакции, отмечавшиеся в течение 24 часов после введения препарата Фесго® или в/в пертузумаба в комбинации с трастузумабом, были отнесены к реакциям, связанным с введением/инфузионным реакциям (см. раздел «Способ применения и дозы» и раздел «Особые указания»).
Реакции, связанные с введением, наблюдались у 0.8% пациентов, получавших препарат Фесго®, инфузионные реакции наблюдались у 10.7% пациентов, получавших в/в пертузумаб и трастузумаб. Большинство системных инфузионных реакций/реакций, связанных с введением, которые отмечались при применении препарата Фесго® или в/в пертузумаба представляли собой озноб, повышение температуры тела или рвоту.
Реакции в месте введения (к которым относилась любая местная реакция, отмечавшаяся в течение 24 часов после введения препарата Фесго®) наблюдались у 14.9% пациентов, получавших препарат Фесго®, и были 1 или 2 степени тяжести. Большинство реакций в месте инъекции при применении препарата Фесго® представляли собой болезненность или покраснение в месте инъекции.
Пертузумаб в/в в комбинации с трастузумабом и химиотерапией
Реакции, связанные с введением препарата, определялись в опорных исследованиях как любое явление, о котором сообщалось как о гиперчувствительности, анафилактической реакции, острой инфузионной реакции или синдроме высвобождения цитокинов, возникшее во время инфузии или в тот же день, что и инфузия. В опорном исследовании CLEOPATRA начальная доза пертузумаба вводилась за день до применения трастузумаба и доцетаксела, что дало возможность изучить реакции, связанные с введением пертузумаба. В первый день, когда проводилось введение только пертузумаба, общая частота инфузионных реакций составила 9.8% в группе плацебо и 13.2% в группе пертузумаба; при этом большинство реакций были легкой или средней степени тяжести. Наиболее распространенными инфузионными реакциями (≥1.0%) в группе пертузумаба были повышение температуры тела, озноб, повышенная утомляемость, головная боль, астения, гиперчувствительность и рвота.
Во время второго цикла, когда все препараты вводились в один день, наиболее частыми инфузионными реакциями (≥1.0%) в группе пертузумаба были повышенная утомляемость, лекарственная гиперчувствительность, дисгевзия, гиперчувствительность, миалгия и рвота (см. раздел «Особые указания»).
В исследованиях неоадъювантной и адъювантной терапии пертузумаб вводился в тот же день, что и другие исследуемые препараты. Инфузионные реакции возникли у 18.6% — 25.0% пациентов в первый день введения пертузумаба (в комбинации с трастузумабом и химиотерапией). Тип и тяжесть реакций соответствовали таковым, которые наблюдались в исследовании CLEOPATRA, при этом большинство реакций были легкой или средней степени тяжести.
Реакции гиперчувствительности/анафилаксии
Препарат Фесго® в комбинации с химиотерапией
В опорном исследовании FEDERICA общая частота реакций гиперчувствительности/ анафилаксии, связанных с применением HER2-таргетной терапии, составила 1.6% в группе препарата Фесго® по сравнению с 1.2% в группе пациентов, получавших в/в пертузумаб и трастузумаб. Ни одно из этих явлений не было 3-4 степени тяжести (NCI-CTCAE версия 4.0) (см. раздел «Особые указания»). У одного пациента была отмечена реакции гиперчувствительности/анафилаксии во время или сразу после введения препарата Фесго® во время первого цикла, которая привела к прекращению терапии (см. раздел «Способ применения и дозы» и «Особые указания»).
Пертузумаб в/в в комбинации с трастузумабом и химиотерапией
В опорном исследовании CLEOPATRA при метастатическом раке молочной железы общая частота явлений гиперчувствительности/анафилаксии по оценке исследователя составила 9.3% в группе плацебо и 11.3% в группе пертузумаба в/в; из данных явлений 2.5% и 2% соответственно были 3-4 степени тяжести по классификации NCI-CTCAE. В общей сложности у 2 пациентов в группе плацебо и у 4 пациентов в группе пертузумаба в/в отмечались явления, которые были расценены исследователем как анафилаксия (см. раздел «Особые указания»).
В целом большинство реакций гиперчувствительности были легкой или средней степени тяжести и разрешались после соответствующего лечения. По результатам анализа реакций гиперчувствительности, при изменении режимов дозирования препаратов, установлено, что явления гиперчувствительности были связаны с инфузиями доцетаксела.
В исследованиях неоадъювантной и адъювантной терапии частота реакций гиперчувствительности/анафилаксии была сходной с таковой, которая наблюдалась в исследовании CLEOPATRA. Случаи анафилаксии зафиксированы у 2 пациентов в исследовании NEOSPHERE, получавших терапию пертузумабом и доцетакселом. По данным исследований TRYPHAENA и APHINITY общая частота реакций гиперчувствительности/анафилаксии была выше при применении пертузумаба в комбинации с трастузумабом, доцетакселом и карбоплатином как в ходе неоадъювантной (13.2%), так и в ходе адъювантной (7.6%) терапии. При этом у 2.6% и 1.3% пациентов соответственно наблюдались реакции 3-4 степени тяжести по классификации NCI-CTCAE.
Фебрильная нейтропения
Препарат Фесго® в комбинации с химиотерапией
В опорном исследовании FEDERICA фебрильная нейтропения наблюдалась у 6.5% пациентов, получавших препарат Фесго®, и у 5.6% пациентов, получавших в/в пертузумаб в комбинации с трастузумабом.
Как и в опорных исследованиях внутривенного введения пертузумаба и трастузумаба, более высокая частота возникновения фебрильной нейтропении наблюдалась среди азиатских пациентов, получавших в/в пертузумаб в комбинации с трастузумабом (13.0%), аналогично частота возникновения фебрильной нейтропении у азиатских пациентов, получавших Фесго®, также была выше (13.7%).
Пертузумаб в/в в комбинации с трастузумабом и химиотерапией
В опорном исследовании CLEOPATRA у большинства пациентов в обеих группах лечения как минимум однократно отмечалась лейкопения (63.0% пациентов в группе пертузумаба и 58.3% пациентов в группе плацебо), в большинстве случаев – нейтропения (см. раздел «Особые указания»). Фебрильная нейтропения возникла у 13.7% пациентов, получавших пертузумаб, и у 7.6% пациентов, получавших плацебо. В обеих группах лечения доля пациентов с фебрильной нейтропенией была самой высокой в первом цикле терапии и снижалась в дальнейшем. Среди пациентов азиатского происхождения в обеих группах лечения наблюдалась повышенная частота фебрильной нейтропении по сравнению с пациентами других рас и из других географических регионов. Среди азиатских пациентов частота возникновения фебрильной нейтропении была выше в группе пертузумаба (25.8%) по сравнению с группой плацебо (11.3%).
В исследовании NEOSPHERE у 8.4% пациентов, получавших неоадъювантную терапию пертузумабом, трастузумабом и доцетакселом, наблюдалась фебрильная нейтропения по сравнению с 7.5% пациентов, получавших трастузумаб и доцетаксел. В исследовании TRYPHAENA фебрильная нейтропения отмечалась у 17.1% пациентов, получавших неоадъювантную терапию пертузумабом + TCH, и у 9.3% пациентов, получавших неоадъювантную терапию пертузумабом, трастузумабом и доцетакселом после терапии FEC. В исследовании TRYPHAENA частота фебрильной нейтропении была выше у пациентов, получивших шесть циклов пертузумаба, по сравнению с пациентами, получившими три цикла пертузумаба, независимо от проводимой химиотерапии. Как и в исследовании CLEOPATRA, в обоих исследованиях неоадъювантной терапии наблюдалась более высокая частота нейтропении и фебрильной нейтропении среди пациентов азиатского происхождения по сравнению с другими пациентами. В исследовании NEOSPHERE у 8.3% азиатских пациентов, получавших неоадъювантное лечение пертузумабом, трастузумабом и доцетакселом, наблюдалась фебрильная нейтропения по сравнению с 4.0% азиатских пациентов, получавших неоадъювантное лечение трастузумабом и доцетакселом.
В исследовании APHINITY фебрильная нейтропения возникла у 12.1% пациентов, получавших пертузумаб, и у 11.1% пациентов, получавших плацебо. Как и в исследованиях CLEOPATRA, TRYPHAENA и NEOSPHERE, в исследовании APHINITY среди пациентов азиатского происхождения, получавших лечение пертузумабом, наблюдалась более высокая частота фебрильной нейтропении по сравнению с пациентами других рас (15.9% пациентов, получавших пертузумаб, и 9.9% пациентов, получавших плацебо).
Диарея
Препарат Фесго® в комбинации с химиотерапией
В опорном исследовании FEDERICA диарея наблюдалась у 61.7% пациентов, получавших препарат Фесго®, и у 59.1% пациентов, получавших в/в пертузумаб и трастузумаб. Диарея ≥3 степени тяжести отмечалась у 7.3% пациентов в группе препарата Фесго® по сравнению с 5.2% в группе пациентов, получавших в/в пертузумаб и трастузумаб. Большинство явлений были 1 или 2 степени тяжести. Наибольшая частота диареи (все степени тяжести) отмечалась в период таргетной терапии и химиотерапии таксанами (57.7% пациентов в группе препарата Фесго® по сравнению с 53.6% пациентов в группе пациентов, получавших в/в пертузумаб и трастузумаб).
Пертузумаб в/в в комбинации с трастузумабом и химиотерапией
В опорном исследовании CLEOPATRA при метастатическом раке молочной железы диарея наблюдалась у 68.4% пациентов, получавших пертузумаб, и у 48.7% пациентов, получавших плацебо. Большинство случаев были легкой или средней степени тяжести и возникали в первые несколько циклов лечения. Частота диареи 3-4 степени тяжести (по классификации NCI-CTCAE) составила 9.3% у пациентов, получавших пертузумаб, по сравнению с 5.1% у пациентов, получавших плацебо. Медиана продолжительности самого длительного эпизода составила 18 дней у пациентов, получавших пертузумаб, и 8 дней у пациентов, получавших плацебо. Диарея хорошо купировалось проактивным назначением противодиарейных препаратов.
В исследовании NEOSPHERE диарея наблюдалась у 45.8% пациентов, получавших неоадъювантное лечение пертузумабом, трастузумабом и доцетакселом, по сравнению с 33.6% пациентов, получавших трастузумаб и доцетаксел. В исследовании TRYPHAENA диарея наблюдалась у 72.3% пациентов, получавших неоадъювантное лечение пертузумабом + TCH, и у 61.4% пациентов, получавших неоадъювантное лечение пертузумабом, трастузумабом и доцетакселом после терапии FEC. В обоих исследованиях большинство явлений были легкой или средней степени тяжести.
В исследовании APHINITY диарея чаще отмечалась у пациентов, получавших пертузумаб, (71.2%) по сравнению с плацебо (45.2%). Диарея ≥3 степени тяжести была зарегистрирована у 9.8% пациентов в группе пертузумаба по сравнению с 3.7% в группе плацебо. Большинство явлений были 1 или 2 степени тяжести. Наибольшая частота диареи (все степени тяжести) отмечалась в период таргетной терапии + химиотерапии таксанами (61.4% пациентов в группе пертузумаба по сравнению с 33.8% пациентов в группе плацебо). После прекращения химиотерапии частота диареи была значительно ниже: 18.1% пациентов в группе пертузумаба по сравнению с 9.2% пациентов в группе плацебо в период после окончания таргетной терапии.
Сыпь
Препарат Фесго® в комбинации с химиотерапией
В опорном исследовании FEDERICA сыпь наблюдалась у 18.1% пациентов, получавших препарат Фесго®, и у 21.8% пациентов, получавших в/в пертузумаб в комбинации с трастузумабом. Большинство случаев сыпи были 1 или 2 степени тяжести.
Пертузумаб в/в в комбинации с трастузумабом и химиотерапией
В опорном исследовании CLEOPATRA при метастатическом раке молочной железы сыпь наблюдалась у 51.7% пациентов, получавших пертузумаб, по сравнению с 38.9% пациентов, получавших плацебо. Большинство явлений были 1 или 2 степени тяжести, возникали в первых двух циклах и купировались стандартными методами терапии, такими как местное или пероральное лечение акне.
В исследовании NEOSPHERE сыпь наблюдалась у 40.2% пациентов, получавших неоадъювантное лечение пертузумабом, трастузумабом и доцетакселом, по сравнению с 29.0% пациентов, получавших трастузумаб и доцетаксел. В исследовании TRYPHAENA сыпь отмечалась у 36.8% пациентов, получавших неоадъювантное лечение пертузумабом + TCH, и у 20.0% пациентов, получавших неоадъювантное лечение пертузумабом, трастузумабом и доцетакселом после терапии FEC. Частота возникновения сыпи была выше у пациентов, получивших шесть циклов пертузумаба, по сравнению с пациентами, получившими три цикла пертузумаба, независимо от проводимой химиотерапии.
В исследовании APHINITY сыпь наблюдалась у 25.8% пациентов, получавших пертузумаб, по сравнению с 20.3% пациентов, получавших плацебо. Большинство случаев сыпи были 1 или 2 степени тяжести.
Отклонения от нормы лабораторных показателей
Препарат Фесго® в комбинации с химиотерапией
В опорном исследовании FEDERICA частота случаев нейтропении 3-4 степени тяжести по классификации NCI-CTCAE (версия 4) была сбалансирована в двух группах лечения (14.5% пациентов, получавших препарат Фесго®, и 13.9% пациентов, получавших в/в пертузумаб и трастузумаб).
Пертузумаб в/в в комбинации с трастузумабом и химиотерапией
В опорном исследовании CLEOPATRA при метастатическом раке молочной железы частота возникновения нейтропении по классификации NCI-CTCAE (версия 3) 3-4 степени тяжести была сбалансирована в двух группах лечения (86.3% пациентов, получавших пертузумаб, и 86.6% пациентов, получавших плацебо, включая 60.7% и 64.8% случаев нейтропении 4 степени тяжести соответственно).
В исследовании NEOSPHERE частота нейтропении по классификации NCI-CTCAE (версия 3) 3-4 степени тяжести составила 74.5% у пациентов, получавших неоадъювантное лечение пертузумабом, трастузумабом и доцетакселом, по сравнению с 84.5% у пациентов, получавших трастузумаб и доцетаксел, включая 50.9% и 60.2% нейтропении 4 степени тяжести соответственно. В исследовании TRYPHAENA частота нейтропении по классификации NCI-CTCAE (версия 3) 3-4 степени тяжести составила 85.3% у пациентов, получавших неоадъювантное лечение пертузумабом + TCH, и 77.0% у пациентов, получавших неоадъювантное лечение пертузумабом, трастузумабом и доцетакселом после терапии FEC, включая 66.7% и 59.5% явлений нейтропении 4 степени тяжести соответственно.
В исследовании APHINITY частота нейтропении по классификации NCI-CTCAE (версия 4) 3-4 степени тяжести составила 40.6% у пациентов, получавших пертузумаб, трастузумаб и химиотерапию, по сравнению с 39.1% у пациентов, получавших плацебо, трастузумаб и химиотерапию, включая 28.3% и 26.5% явлений нейтропении 4 степени тяжести соответственно.
Переход с внутривенного введения пертузумаба и трастузумаба на терапию препаратом Фесго® (и наоборот)
Переход от внутривенного введения пертузумаба и трастузумаба на терапию препаратом Фесго® (и наоборот) хорошо переносился пациентами. Новых или клинически значимых сигналов по безопасности не было выявлено, все возникшие нежелательные явления соответствовали зарегистрированным в исследовании FEDERICA и в предыдущих исследованиях в/в пертузумаба и трастузумаба.
Пациенты пожилого возраста
В целом в исследовании FEDERICA не было выявлено различий в безопасности препарата Фесго® у пациентов в возрасте ≥65 и <65 лет.
Однако в опорных клинических исследованиях в/в пертузумаба в комбинации с трастузумабом частота снижения аппетита, анемии, уменьшения массы тела, астении, дисгевзии, периферической нейропатии, гипомагниемии и диареи была на ≥5% выше у пациентов ≥65 лет (n= 418) по сравнению с пациентами <65 лет (n= 2926).
Имеются ограниченные данные клинических исследований у пациентов >75 лет, получавших терапию препаратом Фесго® или в/в пертузумаб и трастузумаб. Согласно пострегистрационным данным различий в безопасности пертузумаба в комбинации с трастузумабом у пациентов ≥65 и <65 лет нет.
Передозировка
Случаев передозировки препарата Фесго® в клинических исследованиях у человека не отмечалось. Максимальная изученная доза – 1200 мг пертузумаба/600 мг трастузумаба в составе препарата Фесго®.
Взаимодействие с другими лекарственными средствами
Специальных исследований лекарственных взаимодействий не проводилось.
В/в пертузумаб
Дополнительное исследование у 37 пациентов в рамках опорного клинического исследования при метастатическом раке молочной железы не выявило доказательств лекарственного взаимодействия между пертузумабом и трастузумабом и между пертузумабом и доцетакселом. Кроме того, на основании популяционного фармакокинетического анализа не было выявлено клинически значимого фармакокинетического влияния одновременно применяемого доцетаксела или трастузумаба на пертузумаб. Отсутствие лекарственного взаимодействия было подтверждено фармакокинетическими данными из опорных исследований неоадъювантной и адъювантной терапии при раннем раке молочной железы.
Влияние пертузумаба на фармакокинетику одновременно применяемых цитотоксических препаратов, доцетаксела, паклитаксела, гемцитабина, капецитабина, карбоплатина и эрлотиниба оценивали в пяти исследованиях. Доказательств фармакокинетического взаимодействия между пертузумабом и данными препаратами нет. Фармакокинетика пертузумаба в этих исследованиях была сопоставима с таковой при применении в исследованиях в виде монотерапии.
В/в трастузумаб
Специальных исследований лекарственных взаимодействий трастузумаба у человека не проводилось. В клинических исследованиях клинически значимых взаимодействий между трастузумабом и одновременно применяемыми препаратами не отмечалось.
При применении трастузумаба в комбинации с доцетакселом, карбоплатином или анастрозолом, фармакокинетические параметры данных препаратов, включая трастузумаб, не изменялись.
Концентрации паклитаксела и доксорубицина и их основных метаболитов (6-альфа-гидроксипаклитаксел и доксорубицинол) не изменялись в присутствии трастузумаба. Тем не менее, трастузумаб может повысить общую экспозицию одного из метаболитов доксорубицина (7-дезокси-13-дигидродоксорубицинон). Биологическая активность этого метаболита и клиническое значение повышения его экспозиции неизвестны. В присутствии паклитаксела и доксорубицина изменений в концентрации трастузумаба не наблюдалось.
Результаты изучения фармакокинетики капецитабина и цисплатина при использовании в комбинации с трастузумабом или без него предполагают, что экспозиция биологически активных метаболитов капецитабина (например, фторурацил) не изменялась при одновременном применении цисплатина или цисплатина и трастузумаба. Однако были зарегистрированы более высокие концентрации капецитабина и более длительный период его полувыведения при комбинации с трастузумабом. Данные также указывают, что фармакокинетика цисплатина не изменялась при одновременном применении капецитабина или капецитабина в комбинации с трастузумабом.
Особые указания
В медицинской документации пациента следует указывать торговое наименование препарата Фесго® и номер серии.
Дисфункция левого желудочка
На фоне применения препаратов, блокирующих активность HER2, включая пертузумаб и трастузумаб, наблюдалось снижение ФВЛЖ. Частота симптоматической систолической дисфункции левого желудочка (застойная сердечная недостаточность) была выше при применении пертузумаба в комбинации с трастузумабом и химиотерапией по сравнению c применением только трастузумаба и химиотерапии. В ходе адъювантной терапии большинство случаев симптоматической сердечной недостаточности отмечалось у пациентов, получавших химиотерапию на основе антрациклинов (см. раздел «Побочное действие»). У пациентов, ранее получавших антрациклины или лучевую терапию на область грудной клетки, риск снижения ФВЛЖ может быть выше на основании данных исследований в/в пертузумаба в комбинации с трастузумабом и химиотерапией.
Препарат Фесго® и/или в/в пертузумаб и трастузумаб не исследовались у пациентов с исходным значением ФВЛЖ <55% (ранний рак молочной железы) или <50% (метастатический рак молочной железы); застойной сердечной недостаточностью в анамнезе; при состояниях, которые способны нарушать функцию левого желудочка, таких как неконтролируемая артериальная гипертензия, недавно перенесенный инфаркт миокарда, серьезные нарушения сердечного ритма, требующие лекарственной терапии, или предшествующее лечение антрациклинами с кумулятивной дозой доксорубицина или эквивалентного препарата >360 мг/м2.
В/в пертузумаб в комбинации с трастузумабом и химиотерапией не изучались у пациентов с ранее наблюдавшимся снижением ФВЛЖ до значений <50% в ходе адъювантной терапии трастузумабом.
ФВЛЖ следует оценить перед началом лечения препаратом Фесго® и регулярно определять на фоне лечения для того, чтобы убедиться, что ФВЛЖ находится в пределах нормальных значений (см. раздел «Способ применения и дозы»). Если ФВЛЖ снижается (см. раздел «Способ применения и дозы») и не улучшается, или происходит ее дальнейшее снижение после повторной оценки, следует рассмотреть вопрос об отмене препарата Фесго®, если только не будет принято решение, что преимущества его применения для конкретного пациента превосходят риск.
Реакции, связанные с введением/инфузионные реакции
При применении препарата Фесго® отмечались реакции, связанные с введением препарата. К реакциям, связанным с введением, были отнесены любые системные реакции с такими симптомами как лихорадка, озноб, головная боль, вероятно, связанные с высвобождением цитокинов. Данные реакции развивались в течение 24 часов после введения препарата Фесго®.
При введении препарата Фесго® рекомендуется тщательно наблюдать за пациентом на протяжении введения нагрузочной дозы и в течение 30 минут после окончания инъекции, а также на протяжении введения поддерживающей дозы и в течение 15 минут после окончания инъекции.
При развитии клинически значимой реакции, связанной с введением, следует замедлить скорость введения или прервать инъекцию и провести соответствующие лечебные мероприятия. Следует тщательно наблюдать за пациентом и оценивать его состояние до полного разрешения симптомов. У пациентов с тяжелыми реакциями, связанными с введением, следует оценить необходимость полной отмены препарата с учетом степени тяжести наблюдавшейся реакции и характера ответа на лечение, назначенного в связи с нежелательной реакцией. Хотя при терапии препаратом Фесго® летальных исходов в результате реакций, связанных с введением, не наблюдалось, при его применении следует соблюдать осторожность, поскольку при терапии в/в пертузумабом в комбинации с в/в трастузумабом и химиотерапией отмечались инфузионные реакции с летальным исходом.
Реакции гиперчувствительности/анафилаксии
Необходимо тщательно следить за состоянием пациента на предмет развития у него реакций гиперчувствительности.
Хотя при терапии препаратом Фесго® тяжелых реакций гиперчувствительности, включая анафилаксию и явления с летальным исходом, не наблюдалось, следует соблюдать осторожность при его применении, поскольку при терапии в/в пертузумабом в комбинации с трастузумабом и химиотерапией данные реакции отмечались (см. раздел «Побочное действие»). В случае возникновения реакций гиперчувствительности/анафилаксии необходимо незамедлительно начать соответствующие мероприятия и использовать необходимые лекарственные препараты и оборудование для оказания неотложной помощи. Препарат Фесго® противопоказан при наличии у пациента гиперчувствительности к пертузумабу, трастузумабу или к другим компонентам препарата.
Фебрильная нейтропения
У пациентов, получающих терапию в/в пертузумабом, трастузумабом и доцетакселом, повышен риск развития фебрильной нейтропении по сравнению с пациентами, получающими терапию только трастузумабом и доцетакселом, особенно в течение первых 3-х циклов терапии.
При терапии метастатического рака молочной железы минимальные значения числа нейтрофилов были схожи у пациентов, получавших в/в пертузумаб, трастузумаб и доцетаксел, и у пациентов, получавших трастузумаб и доцетаксел.
Таким образом, более высокая частота фебрильной нейтропении у пациентов, получающих в/в пертузумаб, была связана с более высокой частотой мукозита и диареи, в связи с чем следует рассмотреть возможность симптоматического лечения мукозита и диареи. О случаях фебрильной нейтропении после исключения из схемы лечения доцетаксела не сообщалось.
Злоупотребление приемом препарата или лекарственная зависимость
Нет доказательств того, что препарат Фесго® может привести к злоупотреблению его приемом и лекарственной зависимости.
Особые группы пациентов
Пациенты пожилого возраста
В целом профиль эффективности и безопасности препарата Фесго® у пациентов ≥65 и <65 лет не отличался.
Однако, при применении в/в пертузумаба в комбинации с трастузумабом частота развития нежелательных явлений всех степеней тяжести, таких как снижение аппетита, анемия, уменьшение массы тела, астения, дисгевзия, периферическая нейропатия, гипомагниемия и диарея, была, как минимум, на 5% выше у пациентов ≥65 лет по сравнению с пациентами <65 лет (см. раздел «Способ применения и дозы»).
Обращение с неиспользованным препаратом/препаратом с истекшим сроком годности
Попадание лекарственного препарата в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препараты с помощью сточных вод или вместе с бытовыми отходами.
Необходимо строго соблюдать следующие рекомендации по использованию и утилизации шприцев и расходных материалов:
- иглы и шприцы нельзя использовать повторно;
- использованные иглы и шприцы помещают в защищенный от проколов контейнер (емкость).
Уничтожение неиспользованного препарата или расходных материалов должно проводиться в соответствии с локальными требованиями.
Влияние на способность управлять транспортными средствами, механизмами
Препарат Фесго® оказывает небольшое влияние на способность управлять транспортными средствами, механизмами. При терапии препаратом Фесго® могут развиться реакции, связанные с введением, и головокружение (см. разделы «Побочное действие» и «Особые указания»).
Форма выпуска
Раствор для подкожного введения 600 мг + 600 мг/10 мл и 1200 мг + 600 мг/15 мл
По 600 мг + 600 мг/10 мл или 1200 мг + 600 мг/15 мл препарата во флакон бесцветного стекла (гидролитический класс 1 ЕФ), укупоренный пробкой из бутилкаучука, ламинированного фторполимером, обжатый алюминиевым колпачком и закрытый пластмассовой крышкой.
1 флакон с препаратом вместе с инструкцией по применению помещают в картонную пачку.
С целью контроля первого вскрытия на упаковку наносится защитная голографическая наклейка.
Срок годности
18 месяцев.
Не применять по истечении срока годности.
Условия хранения
Хранить при температуре 2-8 °С в картонной пачке для защиты от света.
Хранить в недоступном для детей месте.
Условия отпуска
Отпускают по рецепту.
Владелец регистрационного удостоверения
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Производитель
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Wurmisweg, 4303 Kaiseraugst, Switzerland
Организация, уполномоченная на принятие претензий от потребителя
Претензии потребителей направлять в компанию АО «Рош-Москва» по адресу:
107031, Россия, г. Москва, Трубная площадь, д. 2, помещение 1, этаж 1, комната 42
тел. (495) 229 29 99, факс (495) 229 79 99
или через форму обратной связи на сайте:
www.roche.ru
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Проанализирован клинический случай длительного эффективного применения комбинации «пертузумаб + трастузумаб + доцетаксел» у пациентки c HER2-положительным метастатическим раком молочной железы (РМЖ) (клиническое исследование WO20698 CLEOPATRA). На фоне указанной терапии наблюдался регресс метастазов в печени на 62%. Продолжительность терапии составила 79 месяцев без признаков прогрессирования и с хорошим качеством жизни. Выживаемость без прогрессирования увеличилась на 6,1 месяца (относительный риск (ОР) 0,62; 95%-ный доверительный интервал (ДИ) 0,51–0,75; p < 0,001). Достигнут рекордный выигрыш в общей выживаемости – 15,7 месяца (ОР 0,68; 95% ДИ 0,56–0,84; р = 0,0002). Режим «пертузумаб + трастузумаб + доцетаксел» рекомендован для первой линии терапии HER2-положительного метастатического или местно-распространенного неоперабельного РМЖ.
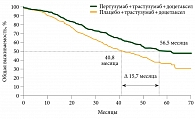
Рис. 1. Медиана общей выживаемости (при медиане наблюдения 50 месяцев)
Рис. 2. Множественные метастазы в печени (компьютерная томография, февраль 2010 г.)
Рис. 3. Уменьшение размеров метастазов в печени (компьютерная томография, апрель 2010 г., через девять недель после начала терапии)
Рис. 4. Максимальное уменьшение размеров метастазов в печени (компьютерная томография, октябрь 2010 г., через 36 недель после начала терапии)
Рис. 5. Эффект терапии сохраняется (компьютерная томография, август 2016 г., через 338 недель после начала терапии)
Введение
В течение последних десятилетий терапия рака молочной железы (РМЖ) претерпела существенные изменения. Определение уровня экспрессии в опухоли рецепторов эстрогенов, рецепторов прогестерона и рецептора HER2 стало рутинной практикой во всех российских онкологических центрах. Гиперэкспрессия HER2 в опухоли выявляется у 20–25% больных РМЖ. Планирование противоопухолевого лечения начинается с определения подтипа РМЖ.
До начала эры таргетной терапии больные HER2-положительным метастатическим РМЖ составляли группу крайне неблагоприятного прогноза. Появление сначала таргетного препарата трастузумаба, затем лапатиниба, а главное – возможность использования этих препаратов в рутинной практике коренным образом изменили судьбу больных HER2-положительным РМЖ [1–3]. Добавление к химиотерапии первой линии трастузумаба позволило значительно увеличить выживаемость без прогрессирования и общую выживаемость при метастатическом HER2-положительном РМЖ [4]. Теперь пациентки с метастатическим РМЖ могут успешно получать одну линию терапии в течение нескольких лет.
К сожалению, часть больных не отвечает на терапию трастузумабом или перестает отвечать на нее спустя время после начала терапии. Примерно у 15% больных HER2-положительным РМЖ отмечается первичная или приобретенная резистентность к трастузумабу [5]. Научный поиск в этой области привел к разработке еще одного анти-HER2-препарата – пертузумаба.
Пертузумаб является гуманизированным моноклональным антителом, связывается с внеклеточным доменом II рецептора HER2 опухоли и препятствует димеризации (образованию пар) рецепторов. Пертузумаб и трастузумаб имеют разные точки приложения на рецепторе HER2.
Семейство рецепторов эпидермального фактора роста хорошо изучено. В него входят четыре трансмембранных тирозинкиназных рецептора: EGFR (erB1), HER2 (erB2), HER3 (erB3) и HER4 (erB4). HER2 отличается от других рецепторов семейства тем, что для его активации не требуется соединения с лигандом. Во внеклеточном домене рецептора HER2 различают четыре части с различными функциями. Пертузумаб блокирует субдомен II, который необходим для димеризации с другими рецепторами семейства erB, а трастузумаб блокирует субдомен IV. Все рецепторы эпидермального фактора роста активны только в виде димеров, причем могут образовываться не только гомодимеры (например, HER2/HER2), но и более активные гетеродимеры. Наиболее активным гетеродимером является HER2/HER3. Считается, что именно образование таких пар рецепторов способствует развитию резистентности к трастузумабу.
Исследования трастузумаба и пертузумаба
В исследовании второй фазы монотерапия пертузумабом при метастатическом РМЖ оказалась эффективной после прогрессирования на фоне трастузумаба [6]. Понимание механизма действия пертузумаба позволяет предположить, что пертузумаб и трастузумаб должны использоваться одновременно для блокирования различных субдоменов рецептора HER2 и прерывания нескольких сигнальных путей. В исследовании второй фазы больные метастатическим РМЖ после прогрессирования на фоне трастузумаба получали комбинацию пертузумаба и трастузумаба. Новая комбинация продемонстрировала высокую эффективность и благоприятный профиль токсичности [7].
Эффективность комбинации пертузумаба и трастузумаба изучали в международном двойном слепом плацебоконтролируемом исследовании III фазы WO20698 CLEOPATRA [8]. Эффективность терапии первой линии HER2-положительного метастатического РМЖ в режиме «пертузумаб + трастузумаб + доцетаксел» сравнивали с таковой комбинации «трастузумаб + доцетаксел». В исследовании участвовали 808 пациенток с HER2-положительным метастатическим РМЖ, ранее не получавших лечения по поводу метастатической болезни. Первичной целью исследования стала выживаемость без прогрессирования по независимой оценке экспертов, дополнительными целями – общая выживаемость и профиль безопасности. Средний возраст больных составил 54 года (27–89 лет). У 48% пациенток опухоль была эстроген- и/или прогестерон-положительной. Неоадъювантную или адъювантную терапию ранее получили 47% больных. Следует отметить, что лишь 10,5% пациенток ранее получили адъювантные режимы, содержавшие трастузумаб. С того момента прошло более 12 месяцев. У 78% больных имели место висцеральные метастазы.
Пертузумаб назначали в нагрузочной дозе 840 мг, затем по 420 мг один раз в три недели, трастузумаб – в стандартном режиме: нагрузочная доза 8 мг/кг, затем по 6 мг/кг один раз в три недели. Доцетаксел в дозе 75 мг/м2 вводили также один раз в три недели. Была предусмотрена эскалация дозы доцетаксела до 100 мг/м2 при отсутствии выраженной гематологической токсичности. После минимум шести (или более до достижения максимального ответа) циклов доцетаксела в отсутствие прогрессирования пациентки продолжали получать только таргетную терапию в режиме «пертузумаб + трастузумаб» или «плацебо + трастузумаб». Уже первый промежуточный анализ при медиане наблюдения 19,3 месяца продемонстрировал высокую эффективность новой комбинации. Объективный ответ (полный + частичный) был достигнут у 80,2% больных в группе двух анти-HER2-препаратов и у 69,3% больных группы «плацебо + трастузумаб» (p = 0,001). По оценке независимых экспертов, медиана выживаемости без прогрессирования в группе больных, получавших комбинацию «пертузумаб + трастузумаб + доцетаксел», достигла 18,5 месяца. В группе трастузумаба и доцетаксела аналогичный показатель составил 12,4 месяца (относительный риск (ОР) 0,62; 95%-ный доверительный интервал (ДИ) 0,51–0,75; p < 0,001). Таким образом, первичная цель исследования была достигнута. В подгруппе больных, которые ранее не получали трастузумаб, различие в показателях медианы выживаемости без прогрессирования было еще значительнее – 21,6 и 12,6 месяца соответственно (ОР 0,6; 95% ДИ 0,43–0,83).
Второй промежуточный анализ был проведен при медиане наблюдения 29,7 месяца в основной группе и 30,1 месяца – в контрольной [9]. Второй промежуточный анализ продемонстрировал впечатляющее преимущество нового режима. Отношение рисков смерти от прогрессирования РМЖ составило 0,66 (р = 0,0008). В связи с этим был проведен кроссовер и больных группы плацебо перевели на более эффективную терапию комбинацией пертузумаба и трастузумаба.
В 2014 г. был представлен финальный анализ данных об общей выживаемости при медиане наблюдения 50 месяцев [10]. Медиана общей выживаемости в группе пертузумаба, трастузумаба и доцетаксела составила 56,5 месяца, в группе плацебо, трастузумаба и доцетаксела – 40,8 месяца (ОР 0,68; 95% ДИ 0,56–0,84; p = 0,0002) (рис. 1). На основании данных исследования CLEOPATRA комбинация «трастузумаб + пертузумаб + доцетаксел» была рекомендована в качестве первой линии терапии метастатического HER2-положительного РМЖ. Наиболее частыми (частота свыше 25% или разница между группами, превышающая 5%) нежелательными явлениями, зарегистрированными в группе пертузумаба, были диарея, сыпь, воспаление слизистых оболочек, головная боль, инфекции верхних дыхательных путей, зуд, фебрильная нейтропения, сухость кожи, мышечные спазмы.
Был проведен подгрупповой анализ в зависимости от возраста пациенток (< 65 лет и > 65 лет) [11]. Интересно, что в подгруппе больных в возрасте 65 лет и старше, которых в исследовании было 15,7%, отмечалась очень высокая эффективность исследуемой комбинации. Более пожилые больные получили в среднем шесть циклов терапии доцетакселом против восьми циклов у больных моложе 65 лет. Объективный ответ у пациенток в возрасте 65 лет и старше зафиксирован в 84% случаев в группе пертузумаба и 75,9% случаев в группе плацебо. Объективный ответ у пациенток моложе 65 лет имел место в 79,5% случаев в группе пертузумаба и 68% случаев в группе плацебо. Медиана выживаемости без прогрессирования для больных в возрасте 65 лет и старше составила 21,6 месяца в группе пертузумаба и 10,4 месяца в группе плацебо (ОР 0,52; 95% ДИ 0,31–0,86). У пациенток моложе 65 лет аналогичный показатель достиг 17,2 месяца в группе пертузумаба и 12,5 месяца в группе плацебо (ОР 0,65; 95% ДИ 0,53–0,80). Интерпретировать полученные данные следует осторожно, поскольку подгруппа пожилых больных была относительно невелика, у них реже отмечались висцеральные метастазы, не так много больных ранее получали трастузумаб. Тем не менее для клинической практики важно, что проведение шести циклов с использованием комбинации «пертузумаб + трастузумаб + доцетаксел» оказалось высокоэффективным. Частота развития нежелательных явлений у пожилых пациенток была несколько выше, чаще развивались диарея, периферическая нейропатия. В то же время влияния на функцию сердечной мышцы не зарегистрировано.
Данные исследования CLEOPATRA показывают, что комбинация «пертузумаб + трастузумаб + доцетаксел» наиболее эффективна в первой линии терапии HER2-положительного метастатического РМЖ.
Клинический случай
Больная Б. 1952 г. р.
Диагноз: рак левой молочной железы T4bN1M0, состояние после комплексного лечения в 2008 г. Прогрессирование в 2009 г. Метастатическое поражение костей и печени (IV клиническая группа).
В марте 2008 г. выявлен рак левой молочной железы T4bN1M0. Гистологическое заключение (трепан-биопсия) – инфильтрирующий рак молочной железы. При иммуногистохимическом исследовании рецепторы эстрогенов – 7, рецепторы прогестерона – 4, HER2-«3+». Было проведено комплексное лечение: четыре цикла неоадъювантной химиотерапии по схеме FAC (фторурацил, доксорубицин, циклофосфамид), радикальная мастэктомия по Маддену слева, послеоперационная лучевая терапия (40–46 Гр), четыре цикла адъювантной химиотерапии по схеме FAC (суммарная доза доксорубицина – 326 мг/м2). Гистологическое исследование послеоперационного материала: инфильтрирующий протоковый рак, первая степень лечебного патоморфоза. Метастаз в одном из 16 лимфоузлов. Адъювантная химиотерапия завершена в ноябре 2008 г. В декабре 2008 г. пациентка начала получать тамоксифен 20 мг/сут.
Прогрессирование заболевания в декабре 2009 г. Выявлен метастаз в правом тазобедренном суставе. Проведена лучевая терапия 25 Гр (по 5 Гр) на эту область. В январе 2010 г. обнаружены метастазы в печени. В феврале 2010 г., по данным компьютерной томографии органов грудной клетки, брюшной полости и малого таза с внутривенным контрастированием, в печени (рис. 2) определены множественные метастатические очаги. Наибольшие из них в S7 – 7 × 6 см, S5/S8 – 6,5 × 4,2 см, S8 – 3,7 × 2,9 см, S3 – 3,1 × 2,2 см, S1 – 4 × 2,3 см. Кроме того, были обнаружены множественные метастазы в костях таза.
С февраля 2010 г. больная стала участницей исследования CLEOPATRA. Пациентке проведено шесть циклов химиотерапии в сочетании с таргетной терапией: пертузумаб назначали в нагрузочной дозе 840 мг, затем по 420 мг один раз в три недели, трастузумаб в нагрузочной дозе 8 мг/кг, затем по 6 мг/кг один раз в три недели, доцетаксел в дозе 75 мг/м2 один раз в три недели. Эскалация дозы доцетаксела не проводилась. При первой оценке через девять недель от начала терапии (после трех циклов) отмечался частичный ответ очагов в печени, очаги уменьшились на 31,6% по критериям RECIST (рис. 3). При оценке ответа на терапию через 18 недель от ее начала эффект продолжал нарастать и регресс измеряемых очагов печени составил 40,3% по сравнению с размерами очагов до лечения. После проведения шести циклов, с июня 2010 г., больная перестала получать доцетаксел и получала только таргетную терапию «пертузумаб + трастузумаб» один раз в три недели. Размеры метастатических очагов печени продолжали уменьшаться. Через 27 недель от начала терапии регресс составил 51%. При проведении компьютерной томографии через 36 недель от начала лечения регресс очагов составил 62,9%: в S7 – 2,9 × 1,6 см, S5/S8 – 5,2 × 2,9 см, S8 – 0,9 × 0,5 см. Очаги в S3 и S1 перестали определяться (рис. 4). В костях таза сохранялась стабилизация. При этом полностью отсутствовал болевой синдром.
Достигнутый в октябре 2010 г. частичный ответ сохраняется по настоящее время (рис. 5). Длительность первой линии терапии на данный момент составляет 79 месяцев. Состояние пациентки в течение терапии удовлетворительное (ECOG 1). Нежелательные явления были связаны только с терапией доцетакселом – нейтропения 3–4-й степени, тошнота 2-й степени, алопеция 2-й степени. С момента окончания терапии доцетакселом нежелательных явлений, связанных с применением трастузумаба или пертузумаба, не зарегистрировано. Гематологической и негематологической токсичности не зафиксировано. Масса тела пациентки остается стабильной. Больная ведет активный образ жизни. Показатели качества жизни, оцениваемые по опроснику FACT-B, стабильны в течение шести лет.
Обсуждение
Рассмотренный клинический случай иллюстрирует высокую эффективность комбинации «пертузумаб + трастузумаб + доцетаксел» в первой линии терапии метастатического HER2-положительного РМЖ. У пациентки имели место множественные висцеральные метастазы, распространенность метастатического процесса послужила показанием к проведению агрессивной химиотерапии в сочетании с таргетной терапией. У больной был определен суррогатный подтип опухоли – люминальный В (HER2-положительный). Стандартной терапией на момент включения пациентки в исследование считалась комбинация таксанов и трастузумаба. Добавление трастузумаба к доцетакселу в исследовании M77001 позволило добиться увеличения частоты общего ответа до 61% по сравнению с 34% в группе монотерапии доцетакселом (p = 0,0002) [4]. Статистически значимым стал выигрыш медианы времени до прогрессирования заболевания: 11,7 месяца в группе трастузумаба и доцетаксела по сравнению с 6,1 месяца в группе доцетаксела (p = 0,0001). Медиана общей выживаемости в группе трастузумаба и доцетаксела составила 31,2 месяца против 22,7 месяца в группе доцетаксела (p = 0,0325). Причем в случае прогрессирования процесса больным группы монотерапии доцетакселом назначали трастузумаб. Из 92 больных, получавших трастузумаб и доцетаксел, общий объективный эффект наблюдался у 62% больных с общей выживаемостью 31,2 месяца. Трехлетний рубеж пережили 30,4% пациенток.
Первичной целью исследования CLEOPATRA была выживаемость без прогрессирования по оценке независимых экспертов. Результаты исследования продемонстрировали cтатистически значимый выигрыш в выживаемости без прогрессирования. Медиана выживаемости без прогрессирования в группе пертузумаба была на 6,1 месяца больше, чем в группе плацебо, – 18,5 и 12,4 месяца соответственно (ОР 0,62; 95% ДИ 0,51–0,75; p < 0,001). Что касается рассмотренного клинического случая, выживаемость без прогрессирования или продолжительность первой линии терапии составила уже 79 месяцев, то есть метастатический РМЖ с висцеральными метастазами стал действительно хроническим заболеванием.
Возраст больной на момент начала терапии составлял 57 лет, у нее не было выявлено каких-либо противопоказаний к проведению химиотерапии. Пациентка относилась к группе больных моложе 65 лет. Частота объективного ответа при назначении пертузумаба у таких больных достигала 79,5%. Частота достижения объективного ответа у больных моложе 65 лет контрольной группы (трастузумаб и доцетаксел) также была достаточно высокой – 68%. Комбинация продемонстрировала высочайшую частоту объективного ответа. Он был получен у каждых четырех из пяти больных. В момент, когда пациентка получила шесть циклов комбинации «пертузумаб + трастузумаб + доцетаксел», регресс измеряемых метастатических очагов в печени по критериям RECIST составил 40,3%. Такую выраженную регрессию исследователи расценили как максимально возможную. При последующих оценках степень выраженности клинического ответа возрастала после того, как химиотерапия доцетакселом была завершена и больная продолжала получать только таргетную терапию. Максимальное уменьшение очагов было достигнуто через 36 недель от начала терапии и составило 62%.
Ряд режимов химиотерапии при метастатическом РМЖ позволяет добиться высокого уровня объективного ответа и увеличения выживаемости без прогрессирования. Но увеличение частоты объективного ответа и выживаемости без прогрессирования зачастую не приводит к увеличению общей выживаемости. В исследовании CLEOPATRA медиана общей выживаемости составила 56,5 месяца. Был получен уникальный рекордный выигрыш в медиане общей выживаемости – 15,7 месяца (p = 0,0002). Профиль токсичности режима «пертузумаб + трастузумаб + доцетаксел» удовлетворительный. Уникальность, несомненно, заключается в том, что после минимум шести циклов с доцетакселом больная переходит на терапию двумя таргетными препаратами, которые не вызывают тяжелой гематологической токсичности, тошноты, алопеции. Режим терапии позволяет больным длительно вести привычный образ жизни, качество жизни не снижается.
Данные исследования CLEOPATRA послужили основанием для регистрации комбинации пертузумаба, трастузумаба и доцетаксела в США, Европе и России в качестве режима первой линии терапии метастатического или местно-распространенного неоперабельного HER2-положительного РМЖ. В России комбинация из двух препаратов пертузумаба и трастузумаба зарегистрирована под названием Бейодайм.
Заключение
На фоне применения комбинации «пертузумаб + трастузумаб + доцетаксел» в исследовании CLEOPATRA были достигнуты рекордная выживаемость без прогрессирования и общая выживаемость. Продолжительность жизни пациентки с момента начала лечения составила 79 месяцев без признаков прогрессирования заболевания. Удовлетворительная переносимость, отсутствие серьезных нежелательных явлений, хорошее качество жизни позволяют проводить длительную непрерывную терапию.
Применение режима «пертузумаб + трастузумаб + доцетаксел» делает HER2-положительный метастатический РМЖ действительно хроническим заболеванием.
Перьета
МНН: Пертузумаб
Производитель: Рош Диагностикс ГмбХ
Анатомо-терапевтическо-химическая классификация: Пертузумаб
Номер регистрации в РК:
№ РК-ЛС-5№021585
Информация о регистрации в РК:
28.09.2020 — 28.09.2030
Информация о реестрах и регистрах
Информация по ценам и ограничения
Предельная цена закупа в РК:
1 301 977 KZT
- Скачать инструкцию медикамента
Торговое название
Перьета
Международное непатентованное название
Пертузумаб
Лекарственная форма
Концентрат для приготовления инфузионного раствора 420 мг/14 мл
Состав
Один флакон содержит
активное вещество — пертузумаб 420 мг,
вспомогательные вещества: L-гистидин, кислота уксусная ледяная, сахароза, полисорбат 20, вода для инъекций
Описание
Прозрачная или опалесцирующая бесцветная или слегка коричневатого цвета жидкость.
Фармакотерапетическая группа
Противоопухолевые препараты. Противоопухолевые препараты другие. Моноклональные антитела. Пертузумаб.
Код АТХ L01ХС13
Фармакологические свойства
Фармакокинетика
Фармакокинетические параметры пертузумаба не зависят от возраста, пола и этнической принадлежности.
Всасывание
Пертузумаб вводится внутривенно. Другие пути введения препарата не изучались.
Распределение
Объем распределения в центральной камере (VC) и периферической камере (VP) составлял 3,11 л и 2,46 л, соответственно.
Метаболизм
Метаболизм пертузумаба не изучался. Как и другие антитела, пертузумаб преимущественно подвергается катаболизму.
Выведение
Клиренс пертузумаба составляет приблизительно 0,239 л/сут, период полувыведения (Т1/2) приблизительно равен 17,2 дням.
Фармакокинетика у особых групп пациентов
Пациенты пожилого и старческого возраста
Специальных исследований фармакокинетики пертузумаба на группах пациентов пожилого и старческого возраста не проводились. Результаты популяционного анализа свидетельствуют о том, что возраст не оказывает влияния на фармакокинетические параметры пертузумаба.
Пациенты с нарушением функции почек
Специальных исследований фармакокинетики пертузумаба на группах пациентов с почечной недостаточность не проводилось. По результатам популяционного анализа почечная недостаточность не оказывает влияния на экспозицию пертузумаба. Однако данные для пациентов с почечной недостаточностью средней и тяжелой степени ограничены.
Фармакодинамика
Пертузумаб – противоопухолевое средство, представляет собой рекомбинантные гуманизированные моноклональные антитела, которые избирательно с отвечающим за димеризацию внеклеточным субдоменом II рецептора эпидермального фактора роста человека 2 типа (HER2). Связывание пертузумаба с субдоменом II блокирует процесс лиганд-зависимой гете-родимеризации HER2 с другими белками семейства HER, включая EGFR (рецептор эпидермального фактора роста человека), HER3 (рецептора эпидермального фактора роста человека 3-го типа) и HER4 (рецептора эпидермального фактора роста человека 4-го типа). Таким образом, пертузумаб ингибирует лиганд-инициированную передачу внутри-клеточных сигналов по двум основным сигнальным путям: путь митоген-активированной протеинкиназы (МАР) и путь фосфоинозитид-3-киназы (PI3K). Угнетение данных сигнальных путей способно привести к остановке роста клеток и апоптозу, соответственно. Кроме того, пертузумаб способствует активации антитело-зависимой клеточной цитотоксичности (АЗКЦ).
Пертузумаб в виде моноагента ингибирует пролиферацию клеток опухоли человека. Показано усиление противоопухолевой активности пертузумаба на моделях ксенотрансплантатов с гиперэкспрессией HER2 при его применении в комбинации с трастузумабом.
Иммуногенность
Примерно у 6,2% пациентов, получавших терапию трастузумабом и доцетакселом, и у 2,8% пациентов, получавших трастузумаб в комбинации с доцетакселом и пертузумабом, были обнаружены антитерапевтические антитела. Однозначная связь образования антител к препарату с развитием анафилактической/ аллергической реакции не была установлена ни у одного из 34 пациентов.
Результаты анализа иммуногенности в значительной степени зависят от различных факторов, таких как чувствительность и специфичность анализа, методология проведения анализа, манипуляции с забранными образцами, время забора образцов, сопутствующие препараты и характер основного заболевания. Исходя из этих соображений, сравнение частоты обнаружения антител к пертузумабу и частоты обнаружения антител к другим биологическим препаратам может оказаться неинформативным.
Показания к применению
Метастазирующий рак или неоперабельный местно-рецидивирующий рак молочной железы (МРМЖ) с опухолевой гиперэкспрессией HER2:
-
в комбинации с трастузумабом и доцетакселом для лечения пациентов, не получавших ранее лечение (анти-HER2 и химиотерапию) по поводу основного заболевания
Неоадъювантная терапия рака молочной железы
-
в составе неоадъювантной химиотерапии в комбинации с трастузумабом и доцетакселом в качестве этапа лечения, включающего фторурацил, эпирубицин и циклофосфамид (схема FEC) или карбоплатин, для местно-распространенного (включая воспалительную форму) рака молочной железы или в случаях, когда размер опухоли превышает 2 см в диаметре.
Способ применения и дозы
Перед началом лечения обязательно надо провести исследование опухоли на HER2-статус. Опухолевая гиперэкспрессия HER2 устанавливается основании 3+ баллов по результатам иммуногистохимического исследования или на основании степени амплификации ≥2.0 по результатам гибридизации in situ (ISH). Следует использовать точные и валидированные методы тестирования.
Препарат Перьета вводят только внутривенно капельно! Не вводить препарат струйно или болюсно!
Метастазирующий рак молочной железы
Нагрузочная доза препарата Перьета составляет 840 мг массы тела в виде 60-минутной внутривенной капельной инфузии, затем через каждые 3 недели введение препарата Перьета повторяют в поддерживающей 420 мг в виде 30-60-минутной инфузии.
При применении в комбинаци с препаратом Перьета нагрузочная доза препарата Герцептин составляет 8 мг/кг массы тела, затем через каждые 3 недели введение Герцептина повторяют в поддерживающей дозе 6 мг/кг массы тела в виде 90-минутной инфузии.
При применении в комбинаци с препаратом Перьета рекомендуемая начальная доза доцетаксела составляет 75 мг/м2 в виде внутривенной инфузии. После чего препарат следует вводить в той же дозе каждые 3 недели. При хорошей переносимости доза доцетаксела может быть увеличена до 100 мг/м2.
Указанные лекарственные средства должны вводиться последовательно. Перьета и трастузумаб могут вводиться в любом порядке. Если пациент получает доцетаксел, то его следует вводить после введения препарата Перьета и трастузумаба. После каждой инфузии препарата Перьета и до начала любого последующего вливания Герцептина или доцетаксела рекомендуется период наблюдения в течение 30-60 минут.
Неоадъювантная терапия рака молочной железы
Перьета, Герцептин и доцетаксел вводятся в порядке, описанном выше, в рамках одной из схем лечения:
-
в течение 3 циклов после схемы FEC
-
в течение 4 циклов перед проведением схемы FEC
-
в течение 6 циклов одновременно с применением карбоплатина (в данном случае увеличение дозы доцетаксела более 75 мг/м2 не рекомендуется).
На сегодняшний день недостаточно данных, на основании которых можно было бы рекомендовать одновременное введение антрациклинов и препарата Перьета.
Длительность лечения
При лечении метастазирующего рака молочной железы лечение следует продолжать до прогрессирования заболевания или до развития непереносимой токсичности.
При неоадъювантной терапии рака молочной железы применение препарата Перьета рекомендовано на срок от 3 до 6 циклов в зависимости от схемы лечения, после чего в адъювантном режиме продолжается назначение Герцептина в сумме до 1 года.
Пропуск в плановом введении
Если пропуск в плановом введении препарата Перьета составил менее 6 недель, следует как можно быстрее ввести препарат в дозе 420 мг в виде 30-60 минутной капельной инфузии, не ожидая следующего планового введения.
Если перерыв во введении препарата Перьета составил 6 недель и более, следует ввести препарат в начальной дозе 840 мг в виде 60 минутной капельной инфузии. Затем продолжить введение препарата в дозе 420 мг каждые 3 недели в виде 30-60 минутной капельной инфузии.
Снижение дозы
Снижение дозы препарат Перьета не рекомендуется.
Препарат Перьета следует отменять в случае отмены препарата Герцептин.
При отмене препарата доцетаксел лечение препаратами Перьета и Герцептин можно продолжать до прогрессирования заболевания или развития непереносимой токсичности.
Снижение дозы препарата Герцептин не рекомендуется.
Рекомендации по модификации дозы доцетаксела – см. в инструкции по медицинскому применению препарата доцетаксел.
Инфузионные реакции
Скорость инфузии следует замедлить или остановить при возникновении у пациента инфузионных реакций. Инфузию следует немедленно прервать при развитии реакции гиперчувствительности (анафилаксии).
Нарушение функции левого желудочка
Введение препаратов Перьета и Герцептин должно быть приостановлено как минимум на 3 недели в следующих случаях:
— снижение фракции выброса левого желудочка (ФВЛЖ) до уровня ниже 40%;
— значение ФВЛЖ 40-45%, однако снижение составляет 10% и более по отношению к показателям наблюдавшимся до лечения.
Возобновить лечение препаратами Перьета и Герцептин можно в случае, если показатель ФВЛЖ восстановится до 45% и более, или же будет составлять 40-45%, при этом снижение по отношению к показателям до лечения будет менее 10%.
Если по данным повторной оценки через 3 недели ФВЛЖ не увеличится или произойдет ее дальнейшее снижение, лечение препаратами Перьета и Герцептин следует отменить, если только польза от их применения в каждом конкретном случае не превосходит риск.
Дозирование у особых групп пациентов
Пациенты пожилого и старческого возраста.
Коррекция дозы у пациентов пожилого и старческого возраста не требуется.
Пациенты детского возраста
Эффективность и безопасность препарата Перьета у пациентов младше 18 лет не изучалась.
Пациенты с нарушением функции почек
У пациентов с почечной недостаточностью легкой и средней степени тяжести коррекция дозы препарата Перьета не требуется.
Пациенты с нарушением функции печени
Эффективность и безопасность препарата Перьета у пациентов с нарушением функции печени не изучались.
Приготовление раствора для инфузий
Внимание: препарат Перьета несовместим с 5% раствором глюкозы!
Разведение в таком растворе приводит к химической и физической нестабильности препарата Перьета. Препарат Перьета нельзя смешивать или разводить вместе с другими лекарственными средствами.
Раствор препарата Перьета совместим с инфузионными пакетами, изготовленными из поливинилхлорида, полиэтилена и полиолефина.
Подготовка препарата к введению должна проводиться в асептических условиях.
Препарата Перьета не содержит антимикробных консервантов. В связи с этим необходимо предпринять меры предосторожности для сохранения стерильности приготовленного раствора для инфузий.
Из флакона (флаконов) следует отобрать весь жидкий концентрат и ввести его в инфузионный пакет с 250 мл 0,9% раствора натрия хлорида. Концентрация готового раствора приблизительно составляет 3,36 мг/мл (840 мг/250 мл) для нагрузочной 1,68 ммл (420 мг/250 мл) для поддерживающей дозы.
Затем инфузионный пакет необходимо осторожно перевернуть для перемешивания раствора, избегая пенообразования. Перед введением препарат следует проверить визуально на предмет отсутствия механических примесей и изменения окраски. Раствор для инфузий вводят тотчас после его приготовления.
В исключительных случаях приготовленный раствор может храниться при температуре 2-8ºС, если приготовление раствора для инфузий происходило в асептических и валидированных условиях. При этом за условия хранения (правила хранения и продолжительность) отвечает специалист, готовивший раствор.
Побочные действия
Безопасность применения препарата Перьета была изучена у более чем 1600 пациентов либо в рамках проведения рандомизированных клинических исследований III фазы, либо в рамках исследования фазы I и II у больных с различными злокачественными новообразованиями, получавших преимущественно препарат Перьета в комбинации с другими противоопухолевыми препаратами. Показатели безопасности препарата Перьета, установленные по результатам исследований фазы I и II в целом соответствовали наблюдениям в ходе исследований III фазы, хотя частота возникновения и наибольшая распространенность нежелательных явлений варьировалась в зависимости от использования пертузумаба в качестве монотерапии или одновременного введения с противоопухолевыми препаратами.
Наиболее распространенными побочными эффектами (≥ 50%), связанными с применением препарата Перьета в комбинации с Герцептином и доцетакселом были диарея, алопеция и нейтропения. Наиболее распространенными (≥ 10%) побочными эффектами 3-4 степени тяжести в соответствии с NCI-CTCAE были нейтропения, фебрильная нейтропения и лейкопения. После прекращения терапии доцетакселом у пациентов, получавших препарат Перьета в комбинации с Герцептином наиболее частыми побочными эффектами были диарея (28,1%), кожная сыпь (18,3%), инфекции верхних дыхательных путей (18,3%), головная боль (17,0%), назофарингит (17,0%), кожный зуд (13,7%), утомляемость (13,4%), астения (13,4%) и артралгия (11,4%).
При назначении препарата Перьета в неоадъювантном режиме в комбинации с Герцептином и доцетакселом наиболее частыми (≥ 50%) побочными эффектами были алопеция и нейтропения. Наиболее распространенным (≥ 10%) побочным эффектом 3-4 степени тяжести в соответствии с NCI-CTCAE была нейтропения.
При назначении препарата Перьета в неоадъювантном режиме в комбинации с Герцептином и доцетакселом в течение 3 циклов, после которых следовали 3 цикла режима FEC, наиболее частыми (≥ 50%) побочными эффектами были диарея, тошнота и алопеция. Наиболее распространенными (≥ 10%) побочными эффектами 3-4 степени тяжести в соответствии с NCI-CTCAE были нейтропения и лейкопения.
При назначении препарата Перьета в неоадъювантном режиме в комбинации с Герцептином, карбоплатином и доцетакселом в течение 6 циклов наиболее частыми (≥ 50%) побочными эффектами были диарея и алопеция. Наиболее распространенными (≥ 10%) побочными эффектами 3-4 степени тяжести в соответствии с NCI-CTCAE были нейтропения, фебрильная нейтропения, анемия, лейкопения и диарея.
Далее представлены суммарные данные по побочным эффектам, наблюдавшимся при применении Перьеты при метастатическом раке молочной железы и в неоадъювантном режиме лечения местно-распространенного, воспалительного и раннего рака молочной железы.
Были использованы следующие категории частоты возникновения: очень часто (≥ 1/10), часто (≥ 1/100 до <1/10), нечасто (≥ 1/1000 до <1/100), редко (≥ 1/10000 к <1/1000), очень редко (<1/10000).
Oчень часто:
-
алопеция, кожная сыпь, поражения ногтевых пластинок
-
периферические отеки
-
нейтропения, анемия, лейкопения, фебрильная нейтропения
-
инфекции верхних дыхательных путей, назофарингит
-
диарея, тошнота, рвота, стоматит, запоры, снижение аппетита
-
утомляемость, астения, повышение температуры тела
-
миагия, артралгия
-
головная боль, нарушение вкусовых ощущений, периферическая нейропатия, бессонница
Часто:
-
кожный зуд, сухость кожи, паронихия
-
одышка, плевральный выпот
-
головокружение, периферическая сенсорная нейропатия
-
дисфункция левого желудочка
-
реакции гиперчувствительности
-
увеличение продукции слезной жидкости.
Нарушение функции левого желудочка
При проведении базового исследования CLEOPATRA частота нарушения функции левого желудочка во время лечения была выше в группе приема плацебо, чем в группе пациентов, получавших Перьета (8,6% и 6,6%, соответственно). Заболеваемость симптоматической систолической дисфункцией левого желудочка была также ниже в группе препарата Перьета (1,8% в группе приема плацебо в сравнении с 1,5% в группе препарата Перьета).
В рамках проведения исследования NEOSPHERE (пациенты получали4 цикла Перьеты в неоадъювантном режиме) частота нарушения функции левого желудочка (во время всего периода лечения) была выше у пациентов, получавших Перьету (7,5%) + Герцептин + доцетаксел, чем в группе приема Герцептина и доцетаксела (1,9%). Был отмечен один случай симптоматической систолической дисфункции левого желудочка у пациентов, получавших препарат Перьета и Герцептина.
В рамках проведения исследования TRYPHAENA частота нарушения функции левого желудочка (во время всего периода лечения) была 8,3% в группе пациентов, получивших сначала Перьету + Герцептин + FEC, после чего пациенты получали Перьету + Герцептин + доцетаксел; 9,3% — в группе пациентов, получивших сначала Перьету + Герцептин + доцетаксел, после чего пациенты получали режим FEC и 6,6% — в группе пациентов, получивших Перьету + Герцептин + карбоплатин.
Частота развития симптоматической систолической дисфункции левого желудочка (застойная сердечная недостаточность) у пациентов, получавших антрациклины и препарат Перьета, и у пациентов, получавших перапарат Перьета в сочетании с другими химиопрепаратами 1,3%.
Инфузионные реакции
В качестве инфузионных реакций в рамках проведения исследования III фазы определяли любое фактическое событие, например, аллергическая реакция, анафилактическая реакция, острая инфузионная реакция или синдром выброса цитокинов, произошедшее непосредственно во время инфузии или в течение дня после инфузии. В исследовании CLEOPATRA начальную дозу препарата Перьета вводили накануне введения Герцептина и доцетаксела для анализа реакций, связанных с приемом препарата. В первый день введения препарата Перьета общая частота инфузионных реакций составила 9,8% у пациентов в группе плацебо и 13,2% у пациентов в группе препарат Перьета, при этом степень реакций варьировалась от легкой до умеренной. Наиболее распространенными инфузионными реакциями (≥ 1,0%) у пациентов, получавших препарат Перьета, были гипертермия, озноб, усталость, головная боль, астения и рвота.
Во время второго цикла, когда все лекарства вводили в один и тот же день, наиболее распространенными инфузионными реакциями (≥ 1,0%) у пациентов, получавших препарат Перьета, были утомляемость, аллергическая реакция на препарат, субъективное расстройство вкуса, гиперчувствительность, миалгия и рвота.
В рамках проведения исследований NEOSPHERE и TRYPHAENA, препарат Перьета вводили в один и тот же день с другими исследуемыми лекарственными средствами. Инфузионные реакции совпадали с наблюдавшимися при проведении исследования CLEOPATRA, при этом степень реакций варьировалась от легкой до умеренной.
Аллергические реакции/анафилаксия
В рамках проведения исследования CLEOPATRA общая частота фактического развития аллергических реакций/анафилаксии составила 9,3% у пациентов, получавших плацебо, и 11,3% у пациентов, получавших препарат Перьета, из которых 2,5% и 2,0% были в соответствии с NCI-CTCAE 3-й и 4-й степени, соответственно. В целом, у 2 пациентов в группе приема плацебо, и у 4 пациентов, получивших препарат Перьета, было отмечено развитие анафилактической реакции. В целом, большинство аллергических реакций были от легкой до умеренной степени тяжести и исчезали по завершении лечения. На основании изменений, внесенных в схему исследуемого лечения, большинство реакций были оценены как вторичные по отношению к введению доцетаксела.
При проведении исследований NEOSPHERE и TRYPHAENA, частота аллергических/анафилактических реакции совпадала с наблюдавшейся в рамках проведения исследования CLEOPATRA. При проведении исследования NEOSPHERE у двух пациентов, получившего препараты Перьета и доцетаксел, было отмечено развитие анафилактической реакции. В исследовании TRYPHAENA общая частота аллергических/анафилактических реакций была самой высокой у пациентов, получавших препарат Перьета и в комбинации с доцетакселом, карборлатином и трастузумабом (13,2%), из которых 2,6% были в соответствии с NCI-CTCAE 3-4 степени тяжести.
Противопоказания
-
гиперчувствительность к пертузумабу и/или вспомогательным ингредиентам препарата
-
детский и подростковый возраст до 18 лет.
Лекарственные взаимодействия
Признаков фармакокинетического взаимодействия между препаратом Перьета и трастузумабом, доцетакселом, гемцитабином, эрлотинибом и капецитабином не обнаружено.
Препарат Перьета несовместим с 5% раствором глюкозы. Разведение в таком растворе приводит к химической и физической нестабильности препарата Перьета. Препарат Перьета нельзя смешивать или разводить вместе с другими лекарственными средствами.
Раствор препарата Перьета совместим с инфузионными пакетами, изготовленными из поливинилхлорида, полиэтилена и полиолефина.
Особые указания
Лечение препаратом Перьета следует проводить только под наблюдением онколога. Замена на другой биологический продукт должна производиться с согласия лечащего врача.
На фоне применения препаратов, блокирующих активность HER2, включая препарат Перьета, наблюдалось снижение фракции выброса левого желудочка. Применение препарата Перьета в комбинации с трастузумабом и доцетакселом не сопровождалось повышением частоты симптоматической систолической дисфункции левого желудочка или снижения ФВЛЖ, по сравнению с применением только трастузумаба и доцетаксела. Однако у пациентов, ранее получавших антрациклины или лучевую терапию на область грудной клетки,
риск снижения ФВЛЖ может быть выше.
При неоадъювантном лечении больных количество случаев дисфункции левого желудочка было больше у пациентов, получавших препарат Перьета, чем в у пациентов, получавших Герцептин и доцетаксел. Увеличение количества случаев снижения ФВЛЖ также наблюдалось у пациентов, получавших препарат Перьета в сочетании с Герцептином и доцетакселом; уровни ФВЛЖ восстановились до показателя более, чем 50%, у всех пациентов.
Безопасность и эффективность применения препарата Перьета не изучены у больных с показателями ФВЛЖ ≤50% перед началом лечения; с сердечной недостаточностью в анамнезе; у больных со снижением показателей ФВЛЖ менее, чем 50% во время предварительной адъювантной терапии с применением Герцептина; у больных, состояние которых могло бы нарушить функцию левого желудочка, например, неконтролируемая артериальная гипертензия, недавний случай инфаркта миокарда, серьезная сердечная аритмия, требующая лечения, или предварительное применение антрациклинов в общем количестве > 360 мг/м2 в виде доксорубицина или его эквивалента.
ФВЛЖ следует оценивать перед началом применения Перьеты и регулярно (каждые 3 месяца при лечении метастатического рака молочной железы и как минимум, каждые 6 недель при неоадъювантной терапии) определять на фоне лечения, чтобы убедиться, что ФВЛЖ находится в переделах нормальных значений, установленных в данном учреждении. Если ФВЛЖ составляет менее 40% или 40-45% при снижении более, чем на 10 пунктов от исходного значения, применения препаратов Перьета и Герцептина следует приостановить. Если после повторной оценки, проведенной приблизительно в течение 3 недель, показатель ФВЛЖ не улучшится или произойдет его дальнейшее снижение, следует рассмотреть вопрос об отмене препаратов Перьета и Герцептин, если только не будет решено, что преимущества их применения для конкретного пациента превосходят риск.
Инфузионные реакции и реакции гиперчувствительности
Назначение препарата Перьета было ассоциировано с развитием инфузионных реакций. При введении препарата Перьета следует тщательно наблюдать за пациентом на протяжении первой инфузии и в течение 60 мин после ее окончания, а также на протяжении последующих инфузий и в течение 30 мин после их окончания. При развитии клинически значимой инфузионной реакции следует замедлить скорость инфузии или прервать ее и провести соответствующие лечебные мероприятия. Тщательное наблюдение за пациентом и оценка его состояния рекомендуются до полного разрешения симптомов. У пациентов с тяжелыми инфузионными реакциями следует оценить необходимость полной отмены препарата. Клиническая оценка должна основываться на тяжести развившейся реакции, ответе на применяемую терапию.
Следует тщательно наблюдать за пациентами на предмет реакций гиперчувствительности. В клинических исследованиях наблюдались случаи развития тяжелых реакций гиперчувствительности, включая анафилактические реакции. Лекарственные средства для лечения подобных состояний, а также реанимационное оборудование должно быть в наличии для немедленного использования при необходимости.
Беременность и лактация
Следует избегать назначения Перьеты в период беременности, кроме как в случаях, когда потенциальная польза для матери превышает риск для плода. Женщинам детородного возраста во время лечения препаратом Перьета и, как минимум, в течение 6 месяцев после окончания лечения необходимо использовать надежные методы контрацепции.
В случае наступления беременности необходимо предупредить женщину о возможности вредного воздействия на плод.
Поскольку IgG секретируется с грудным молоком, а вероятные воздействия на организм ребенка неизвестны, должно быть принято решение о прекращении грудного вскармливания или о прекращении назначения препарата, принимая во внимание пользу для здоровья матери и период полувыведения претузумаба.
Особенности влияния лекарственного средства на способность управлять транспортным средством или потенциально опасными механизмами
Учитывая имеющиеся побочные действия препарата Перьета, пациентам, получающим инфузии препарата, следует соблюдать осторожность при управлении автомобилем и выполнении работ, требующих повышенной концентрации внимания.
Передозировка
В проведенных клинических исследованиях случаев передозировки препарата не наблюдалось. Дозы выше 25 мкг массы тела не изучались.
В случае передозировки необходимо тщательное наблюдение за пациентами с целью обнаружения симптомов нежелательных явлений и назначения соответствующего симптоматического лечения.
Форма выпуска и упаковка
По 420 мг/14 мл препарата во флаконы бесцветного стекла типа 1, укупоренный резиновой пробкой с покрытием из фтористого полимера и алюминиевым колпачком с пластиковой крышкой «flip-off».
По 1 флакону вместе с инструкцией по медицинскому применению на государ-ственном и русском языках помещают в картонную пачку.
Условия хранения
Хранить в оригинальной упаковке при температуре от 2 до 8 °С. Не замораживать.
Хранить в недоступном для детей месте!
Срок хранения
2 года
Не применять по истечении срока годности, указанного на упаковке
Условия отпуска из аптек
По рецепту
Производитель
Рош Диагностикс ГмбХ., Маннхайм, Германия
Roche Diagnostics GmbH, Sandhofer Strasse 116, 68305 Mannheim, Germany
Владелец регистрационного удостоверения
Ф. Хоффманн-Ля Рош Лтд., Базель, Швейцария
Упаковщик
Ф. Хоффманн-Ля Рош Лтд., Кайзераугст, Швейцария
F. Hoffmann-La Roche Ltd, Wurmisweg СН-4303 Kaiseraugst, Switzerland
Адрес организации, принимающей на территории Республики Казахстан претензии от потребителей по качеству товара и ответственной за фармаконадзор:
ТОО «Рош Казахстан»
050000, Алматы
ул. Кунаева, д. 77, Бизнес-центр «Park View Office Tower», 15 этаж
e-mail: kz.safety@roche.com, kz.quality@roche.com
| 252098531477976418_ru.doc | 135 кб |
| 910508651477977616_kz.doc | 160 кб |
Отправить прикрепленные файлы на почту
Национальный центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники